7.1 Introduction
Today, the use of biomass is considered a promising way to diminish negative environmental impact. Moreover, in some future scenarios, renewable raw materials are thought to be able to replace finite mineral-oil-based raw materials before 2050 [1]. This means that new synthetic routes, which should desirably adhere to the principles of green chemistry [2], need to be developed for the production of chemicals.
Lignocellulosic biomass, as a renewable source of energy and chemicals, has attracted a lot of attention recently [3-10]. Wood biomass consists of cellulose (40–50%), lignin (3–10%), hemicelluloses (15–30%) and a variety of extractives (1–10%). Cellulose is a linear polymer of D-glucopyranose and can contain up to 10,000 units (C6H10O5), connected by glycosidic ether bonds, while the molecular mass for hemicelluloses is lower. Hemicelluloses have a more heterogeneous structure than cellulose, consisting mainly of five-carbon (xylose, arabinose) and six-carbon sugars (galactose, glucose and mannose). Contrary to cellulose lignin is a coniferyl alcohol polymer with coumaryl, coniferyl and sinapyl alcohols as monomers, which are heavily cross-linked, leading to complex structures of large lignin molecules [11].
Chemical treatment of lignocellulosic biomass in general, and wood in particular, can have several targets. One of the options is delignification of the biomass leading to cellulose and some residual hemicelluloses, which are further applied in the production of paper or board, or derivatives of cellulose. Thermal (or catalytic) treatment of biomass, e.g., thermal or catalytic pyrolysis, is a route to bio-based synthesis gas and biofuels [12]. Depolymerization results in the formation of low-molecular-mass components (sugars, phenols, furfural, various aromatic and aliphatic hydrocarbons, etc.), e.g., unique building blocks for further chemical synthesis.
Wood biomass contains many valuable raw materials for producing fine and specialty chemicals (Figure 7.1). These raw materials are carbohydrates, fatty acids, terpenoids and polyphenols, such as stilbenes, lignans, flavonoids and tannins. Some of them can be exuded directly from living trees, while others are extracted and purified via chemical methods.
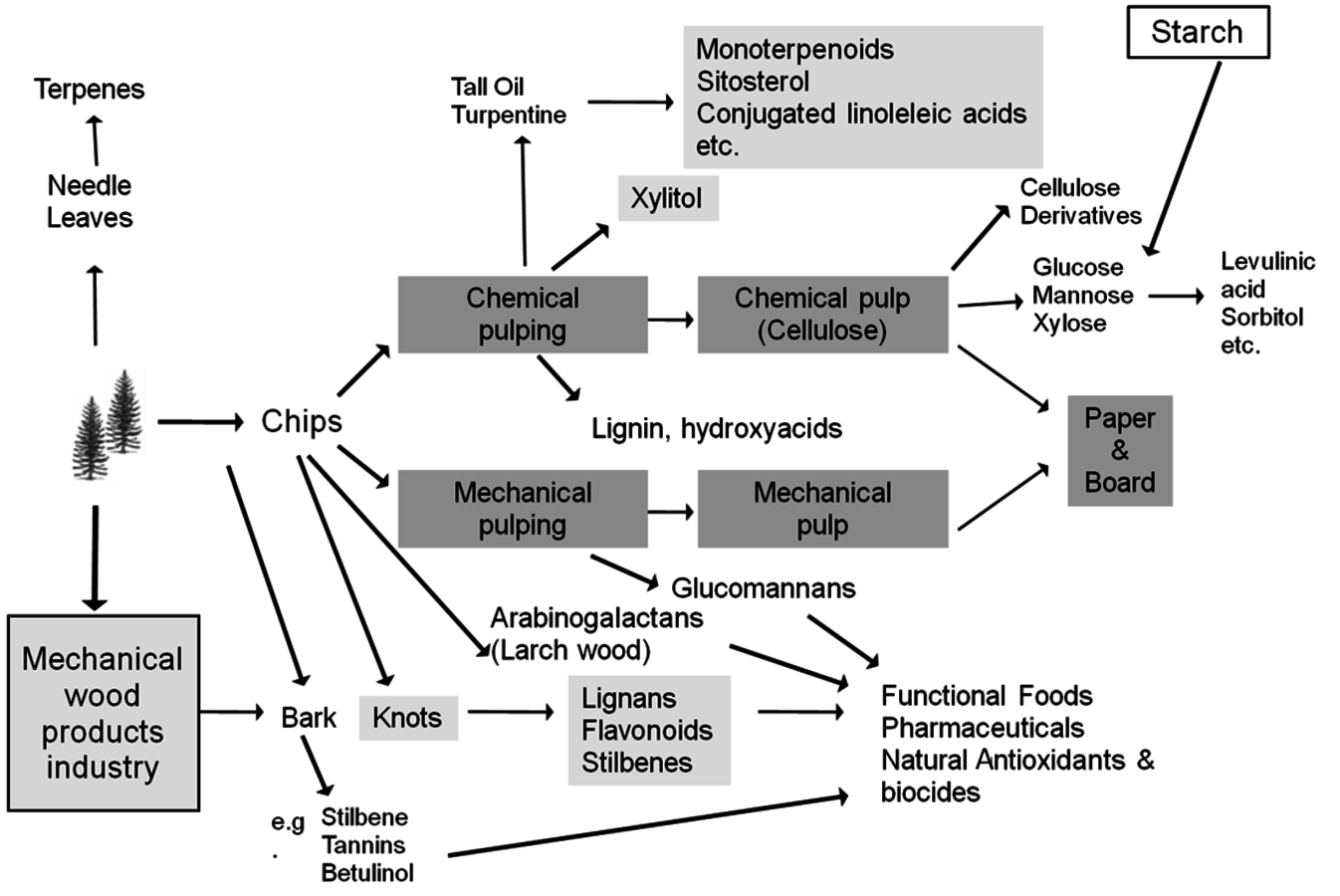
Fig. 7.1: Chemical by-products from the forest industry
In this context, applications of catalytic reagents, which are superior to stoichiometric reagents producing stoichiometric amounts of wastes, are worth mentioning. Well-known benefits in using heterogeneous catalysts are associated with easy catalyst separation, regeneration and reuse, as well as relatively low prices compared to homogeneous catalysts. The research regarding catalytic transformations of different wood-derived compounds is currently very active [13].
Because of the complexity associated with the processing of biomass per se or the transformation of biomass-derived chemicals, in-depth chemical analysis of all components and their reactions is difficult to perform. Therefore, most analytical methods will be a result of a compromise between information depth and available resources. It is also obvious that in industrial processes only a limited number of rather fast analytical methods could be utilized since a large number of samples should be processed.
To have in-depth and molecular-level understanding of the chemical reactions occurring during the transformation of biomass not only advanced analytical methods are required, but additionally, a broad spectrum of these methods needs to be applied. Let us consider, for example, the catalytic conversion of cellulose [14-17] in the presence of hydrogen leading to sugar alcohols. During such a depolymerization reaction not only the concentration of carbohydrates and other products in the liquid phase should be measured, but also the crystallinity of cellulose, its morphology, molecular mass distribution and presence of sugar oligomers. The analysis is even more complicated if in this reaction wood is used directly instead of cellulose.
Analytical techniques have made a tremendous progress in recent years giving a possibility to utilize a wide range of modern instrumental methods, including advanced chromatography, microscopy and spectroscopy. It is apparently clear that all the methods currently available cannot be treated in this review, thus a rational selection of them was done by the authors based on their experience, with an understanding that it might not cover all the analytical methods presently utilized in catalytic transformations of biomass-derived chemicals, but focuses mostly on chromatography.
7.2 Analytical Objectives
Any planning of analytical procedures should be based on the goals and scope of the study. The following critical steps in an analytical process can be listed: problem definition and formulation of analytical objectives; set-up of an analytical plan; sampling; sample transport and storage; sample pretreatment; analytical determination; data calculation; evaluation of results to see if the objectives are achieved.
It is apparently clear from this list that the actual analytical determination is just one step among the others and sometimes could not even be the crucial one. Moreover, preparation of the samples, pretreatment and evaluation of data could be more demanding or at least time-consuming. Since in catalytic transformation of lignocellulosic biomass often wood or various streams from pulping are used as raw materials, a special attention should be devoted to sampling. Inappropriate sampling could undermine the value of the whole study, therefore it should be carefully planned. Sampling and sample storage is important since samples may be altered or destroyed due to temperature, light, presence of oxygen, humidity, enzymes or microbes (bacteria, fungi, etc.). For instance, enzymatic and microbiological attack can happen for samples of fresh wood, wet pulp and paper, sludge, process waters and effluents, while polyunsaturated extractives like abietic acid could be subjected to oxidation. Storage in a frozen state (< -20 °C) gives structural changes in wet, solid materials and physicochemical changes in process waters. Biocides could be added to preserve moist samples. The drying of samples could bring a risk of oxidation, therefore freeze-drying is usually recommended. The latter works by freezing the material and then reducing the surrounding pressure to allow the frozen water in the material to sublime directly from the solid phase to the gas phase.
7.3 Basic Analytical Methods
7.3.1 Chromatography
Chromatographic and spectroscopic methods are widely used today for analytical purposes. Chromatographic techniques are applied not only for off-line analysis, but also for the on-line determination of minute amounts, as well as large-scale preparative separations. In fact, not only monomers, but also polymers and oligomers can be separated by chromatography, although in the former case it is essentially a group separation. There are several forms of chromatography using different mobile and stationary phases, with the two main forms of instrumental chromatography being liquid (LC) and gas chromatography (GC). According to IUPAC definition, chromatography is a physical method of separation in which the components to be separated are distributed between two phases, one of which is stationary, while the other (mobile) moves in a definite direction.
7.3.2 Gas Chromatography
When relating gas chromatography to catalytic transformations of biomass, it can be stated that GC (Figure 7.2) provides qualitative and quantitative determination of organic components such as extractives, hemicellulose building blocks, organic acids, etc. The derivatized and vaporized products are introduced to the column for separation and identified in a detector, whose response is recorded as a chromatogram. Capillary columns made of fused silica with a stationary phase as a thin film of liquid or gum polymer on the inside of the tube are mainly used. The most commonly utilized stationary phases are siloxane polymer gums with different substituents providing different polarity. The polymers are usually cross-linked in the column by photolytic or free-radical reactions, bringing strength to the polymer films. Wall-coated open-tubular columns with a liquid phase coated directly on the inner walls, as well as support-coated open-tubular columns are applied. In the latter case a stationary phase is coated on fine particles deposited on the inner walls. Among non-polar columns, HP-1, DB-1, etc., based on dimethyl, polysiloxane could be mentioned. HP-5 with 95% dimethyl polysiloxane and 5% phenyl groups is slightly more polar. Still more polar columns employ polyoxyethylene or polyester liquid phases.
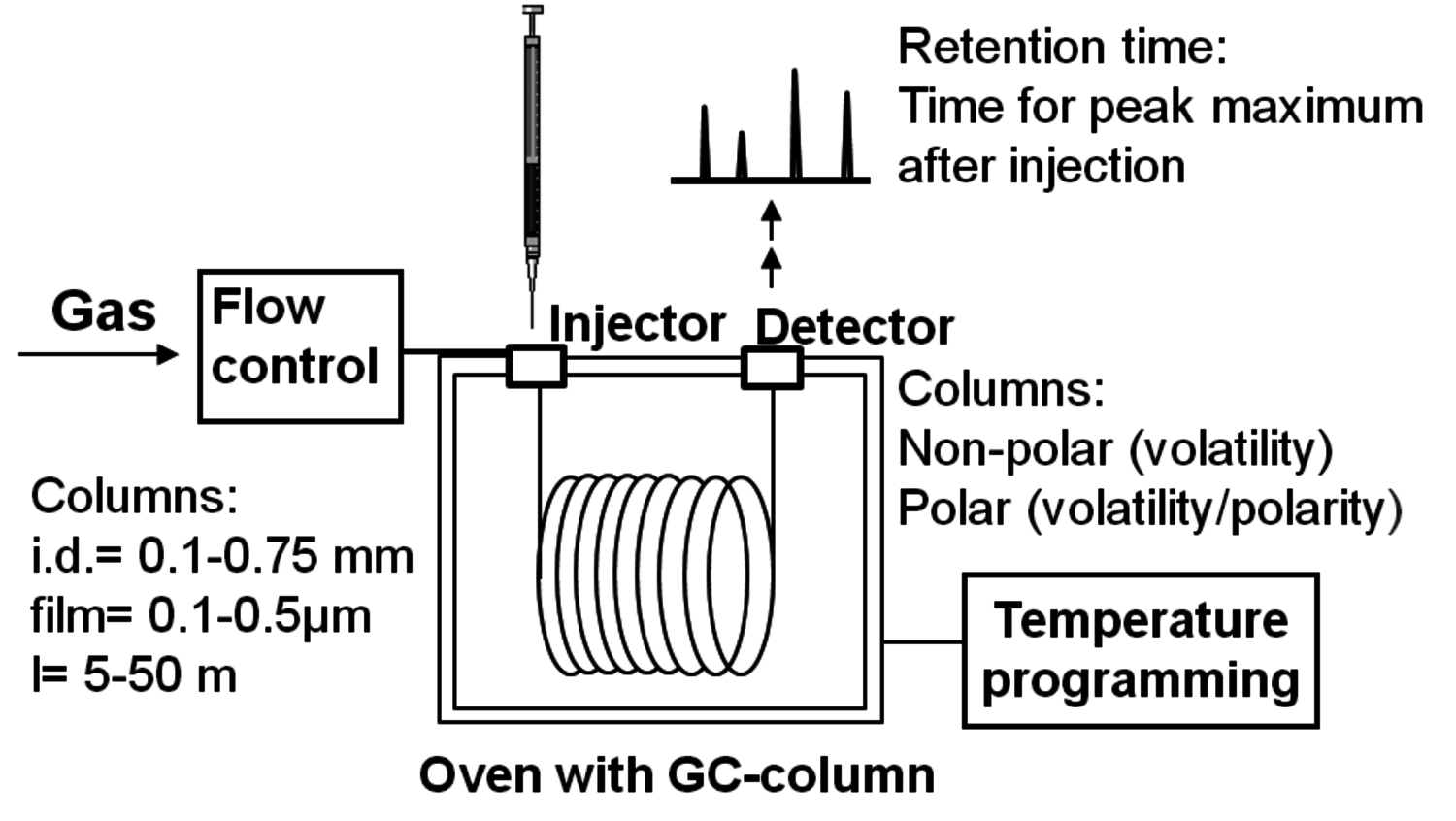
Fig. 7.2: The schematic of gas chromatography
Capillary columns are available in a wide range of internal diameters, lengths and liquid film thicknesses (Figure 7.2). Although longer columns provide better separation, they have an increased analysis time which is usually undesired. In addition, longer columns lead to higher pressure and thus to problems with the injection. Columns with thicker films have higher capacity, but usually require higher temperature, while thin-film columns are suited for large molecules with low volatility. In principle, analysis of components with up to 60 carbon atoms is possible.
Different types of injection systems are used in GC. Split mode, where the injected material after evaporation is split between the column and an outlet, affords rapid volatilization and homogeneous mixing with the carrier gas. Most of the sample will pass out through the split vent and only a small proportion will flow into the column. Splitless systems provide a more reliable quantification allowing analysis of even such high-molecular mass compounds as triglycerides and steryl esters. Flame ionization detectors, which are of destructive nature, have high sensitivity to hydrocarbons, but are not able to detect water. On-line coupling of capillary columns with mass spectrometers is routine nowadays and enables convenient structure identification.
An important but sometimes forgotten issue is the fact that the sensitivity for different compounds is varying for a detector; thus, different peak areas are in proportion to the weight concentration. Knowledge of response factors is therefore necessary and calibration for components especially with various functional groups should be properly done. Commonly, internal standard compounds are applied, e.g., compounds which are not present in the sample itself are purposely added. Chemically they should be similar to the sample compounds with close retention time, however, with no peak overlapping (Figure 7.3).
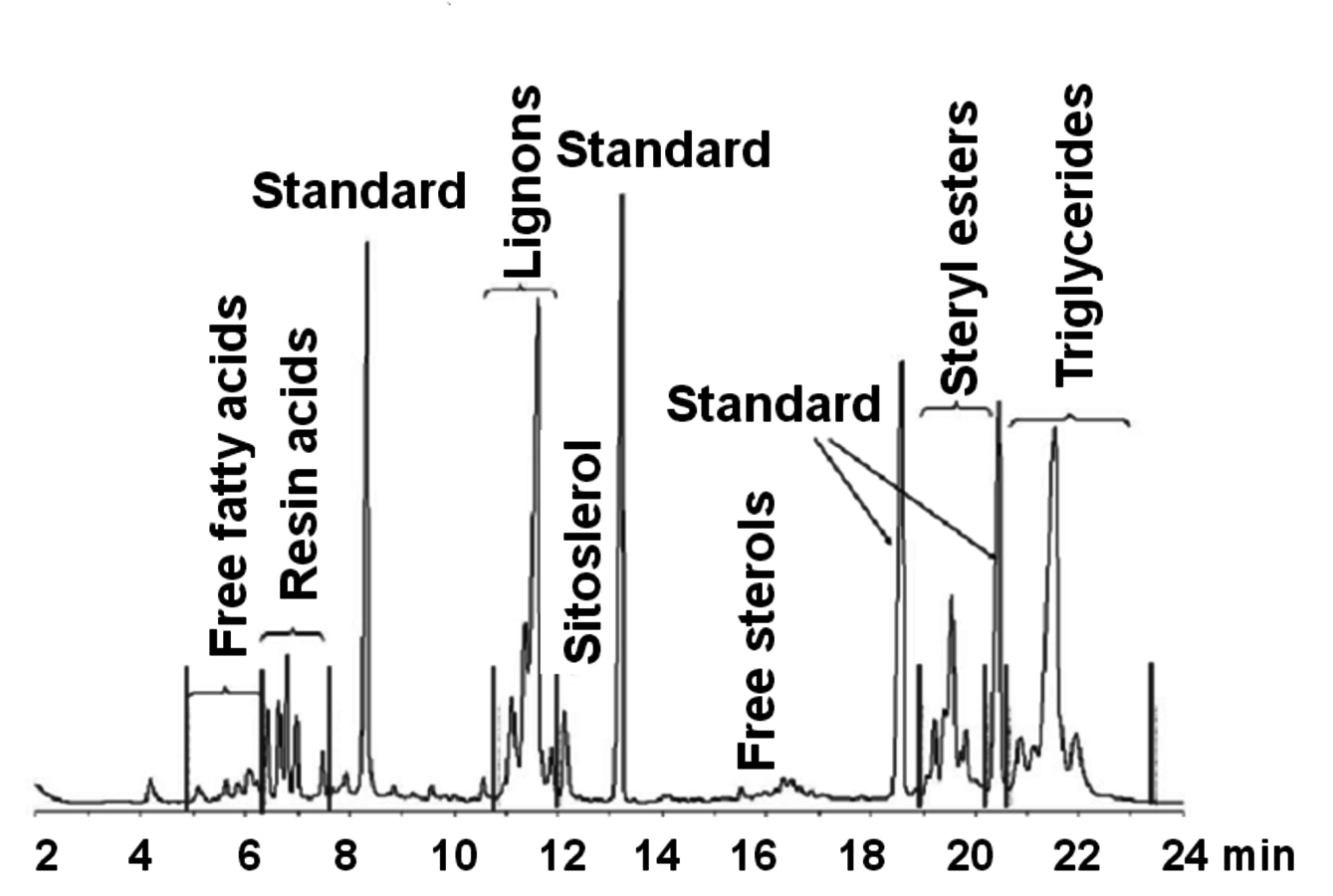
Fig. 7.3: Example of a gas chromatogram on a short column with four standards added to a sample (spruce wood extract) [18]
In addition to such advantages of GC as accurate quantification based on internal standards, a possibility to be combined with a mass spectrometer and complete automation regarding injection and analytical runs, the very high resolution should also be mentioned. On the other hand only molecules up to about 1000 mass units can be analyzed, as they should be stable at high temperatures. Therefore, sometimes samples should be processed before the analysis. The last point is important for polar compounds, like for example acids, which should be derivatized. GC and GC-MS analysis in the vapour phase require volatile derivatives that do not adsorb onto the column wall. Different derivatizations for different substances are recommended, e.g., silylation or methylation for extractives, methanolysis and silylation for carbohydrates. Silyl derivatives of R-O-Si(CH3)3 type containing a trimethylsilyl group (TMS) are formed by the displacement of the active proton in -OH, -NH and -SH groups. Thus, protic sites are blocked, which decreases dipole-dipole interactions and increases volatility. Common silylation reagents are listed in Figure 7.4.
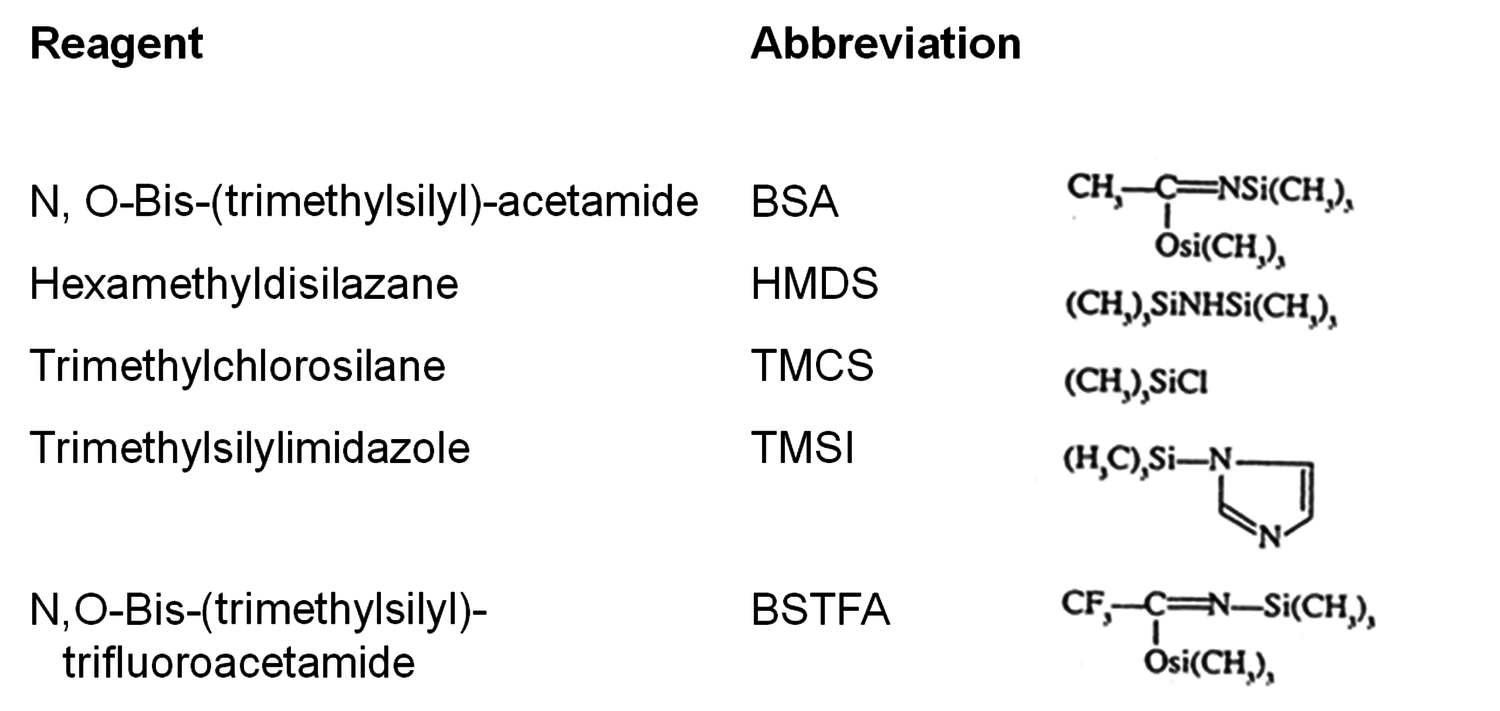
Fig. 7.4: Silylation agents
Methylation relies on the following reactions: utilization of diazometane (CH2N2): R-COOH + CH2N2 = RCOOMe + N2; acid-catalyzed esterification: R-COOH + R´OH => RCOOR´, as well as on-column esterification using tetra- methyl ammonium salts R-COOH + N+Me4OH- => RCOOMe.
One of the variants of GC is associated with coupling pyrolysis to it (Figure 7.5). In this arrangement the sample is thermally degraded in an inert atmosphere. The degradation products are introduced to GC or GC-MS for separation and identification allowing qualitative and quantitative determination of semi-volatile and non-volatile components, such as extractives, polymers, paper chemicals, and lignin, etc.
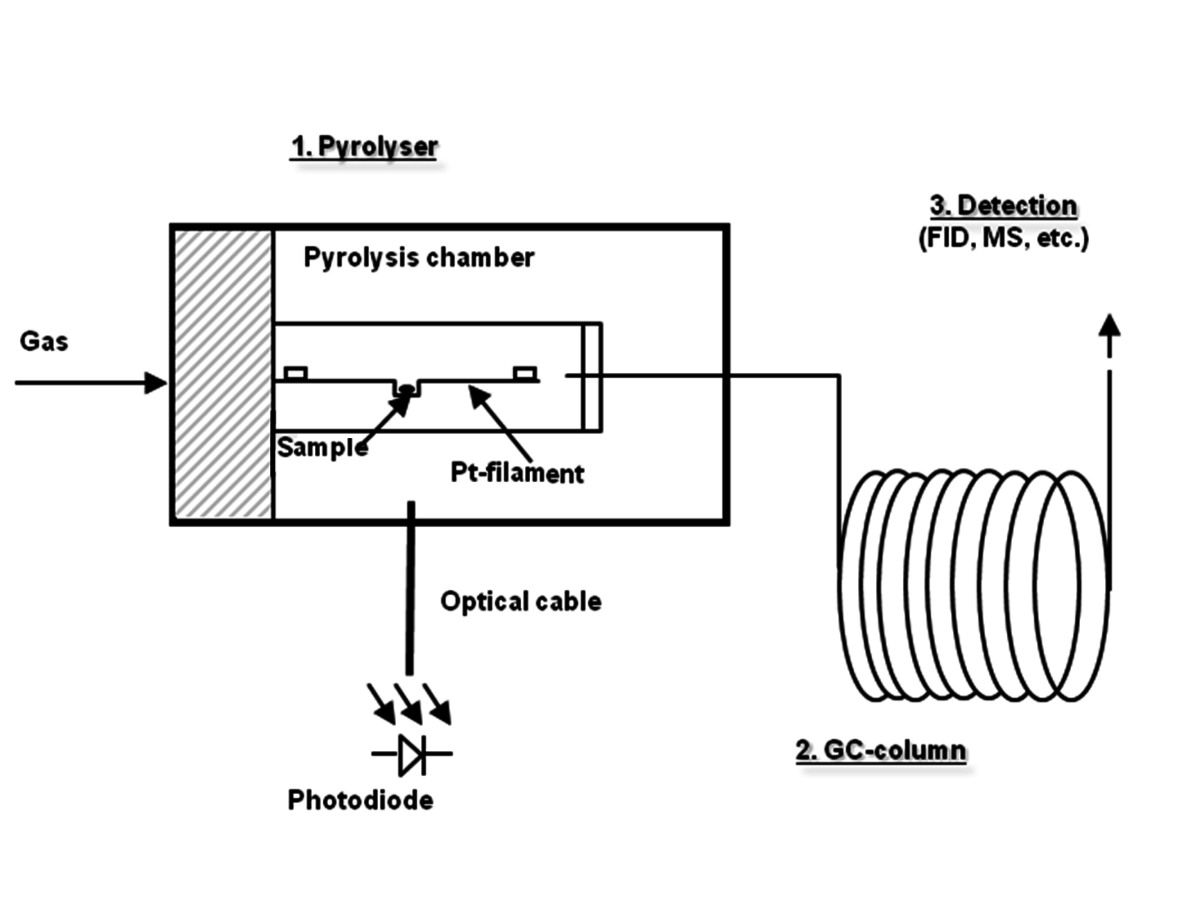
Fig. 7.5: Pyrolysis GC
7.3.3 Liquid Chromatography
These chromatographic methods use liquids such as water or organic solvents as the mobile phase. Silica or organic polymers as well as anion-exchange resins are used as stationary phase. Separation is performed either at atmospheric pressure or at high pressure generated by pumps. The last version is often called high-performance liquid chromatography (HPLC) with solvent velocity controlled by high-pressure pumps, giving a constant flow rate of the solvents. Solvents are used not only as single solvents but they can also be mixed in programmed proportions. In fact, even gradient elution could be applied with increasing amounts of one solvent added to another, creating a continuous gradient and allowing a sufficiently rapid elution of all components.
The most commonly used columns contain small silica particles (3–10 μm) coated with a nonpolar monomolecular layer.
For lipophilic (low-polar) compounds the mobile phase is an organic solvent, while reversed phase HPLC employs mixtures of water and acetonitrile or water and methanol as eluents and is applied for non-ionized compounds soluble in polar solvents. As examples, such columns (Figure 7.6) could be mentioned as Agilent Zorbax SB-Aq (4.6×250 mm, 5 µm) allowing the use of highly aqueous mobile phases working in a pH range from 1 to 8 and affording reproducible retention and resolution for polar compounds. Another example is HypercarbTM (4.6×100 mm, 5 µm) with 100% porous graphitic carbon as a stationary phase, which operates in the pH range 0–14 and can resolve highly polar compounds with closely related structures (e.g., geometric isomers, diastereomers, oligosaccharides). CarboPac PA1 (polymer based) column can be used in mono-, oligo- and polysaccharide analysis by high-performance anion-exchange chromatography combined at high pH with pulsed amperometric detection.
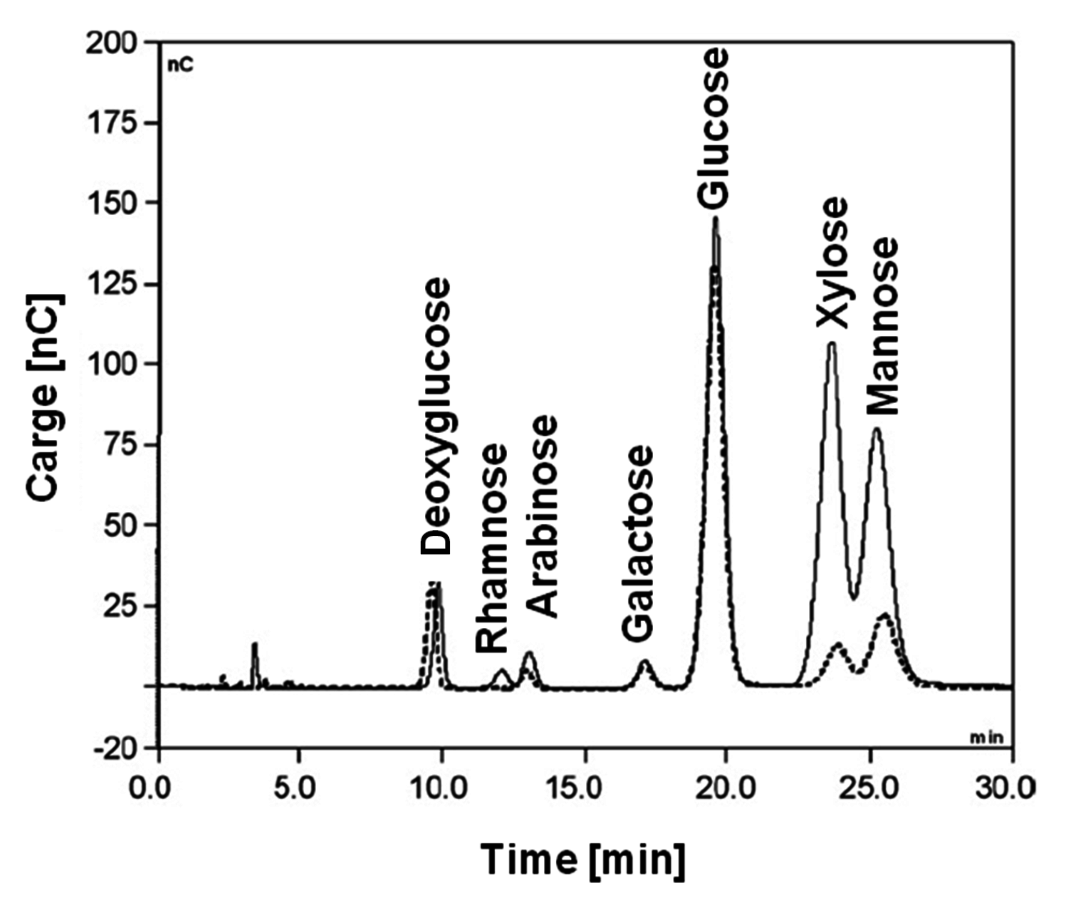
Fig. 7.6: Separation of acids and sugars by HPLC using CarboPac PA1 [19]
UV-Vis (Figure 7.7) and diode-array detectors enabling recording of UV-Vis spectra, for example every second, are common nowadays. They can be used for the analysis of conjugated and aromatic compounds, such as phenols. Another popular detector is based on refractive index (RI) monitoring and is well suited, for example, for carbohydrates. High-performance anion-exchange chromatography with pulsed amperometric detection is a common technique for analyzing sugars in wood and pulp hydrolysates.
Another important form of HPLC is size-exclusion chromatography (Figure 7.8), which is widely applied for the determination of molecular-mass distributions of dissolved lignin and hemicelluloses, and even for cellulose dissolved in ionic liquids. The same method can be used for the analysis of extractives and their derivatives, for instance dimers and trimers of fatty acids [20]. In SEC, solutes in the mobile phase (for example THF) are separated according to their molecular size. Smaller molecules penetrate far into the porous column packing material and thus elute later than larger ones.

Fig. 7.7: A view of LC-UV
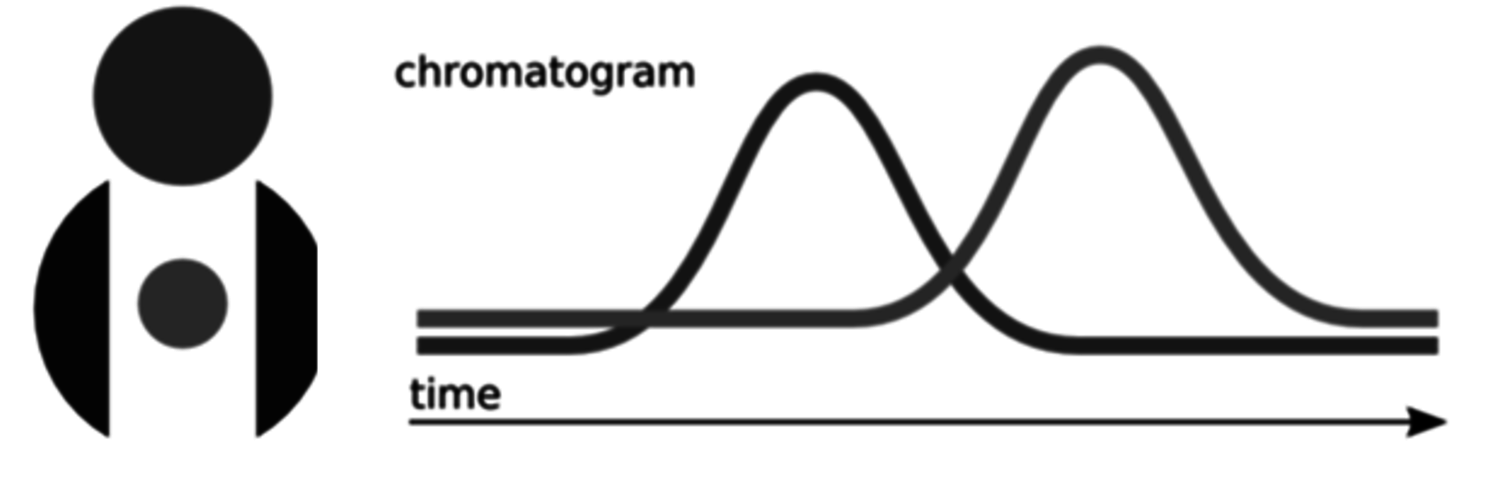
Fig. 7.8: Size-exclusion chromatography
The non-destructive character as well as the absence of derivatization could be mentioned among the advantages of LC. This technique can handle both small and large amounts and it can be used also for preparative isolation of compounds from mixtures. Contrary to GC there are almost no, or at least much fewer, limitations in terms of the molecular size. In addition, LC can be combined with mass spectrometry, once again without derivatization. Thermally unstable and polar compounds can thus be analyzed as such, and the molecular mass in triple quadrupole or ion-trap LC-MS can be up to m/z 3000, while time-of-flight versions allow even up to 16,000.
LC-MS provides better sensitivity and selectivity than GC-MS and is excellent for the quantification of selected substances in complex mixtures. On the other hand, this technique is not very suitable for rapid and reliable identification of unknown compounds mainly because fragmentation is sparse as the conditions of ionization are mild. Furthermore, spectra libraries enabling identification are not available. Other shortcomings of LC-MS are the rather low sensitivity of the detectors for certain compounds. Moreover, it may be difficult to obtain constant pressure, which in turn influences retention; clean, degassed solvents are needed and, finally, it might be challenging to find the optimum solvent mixture.
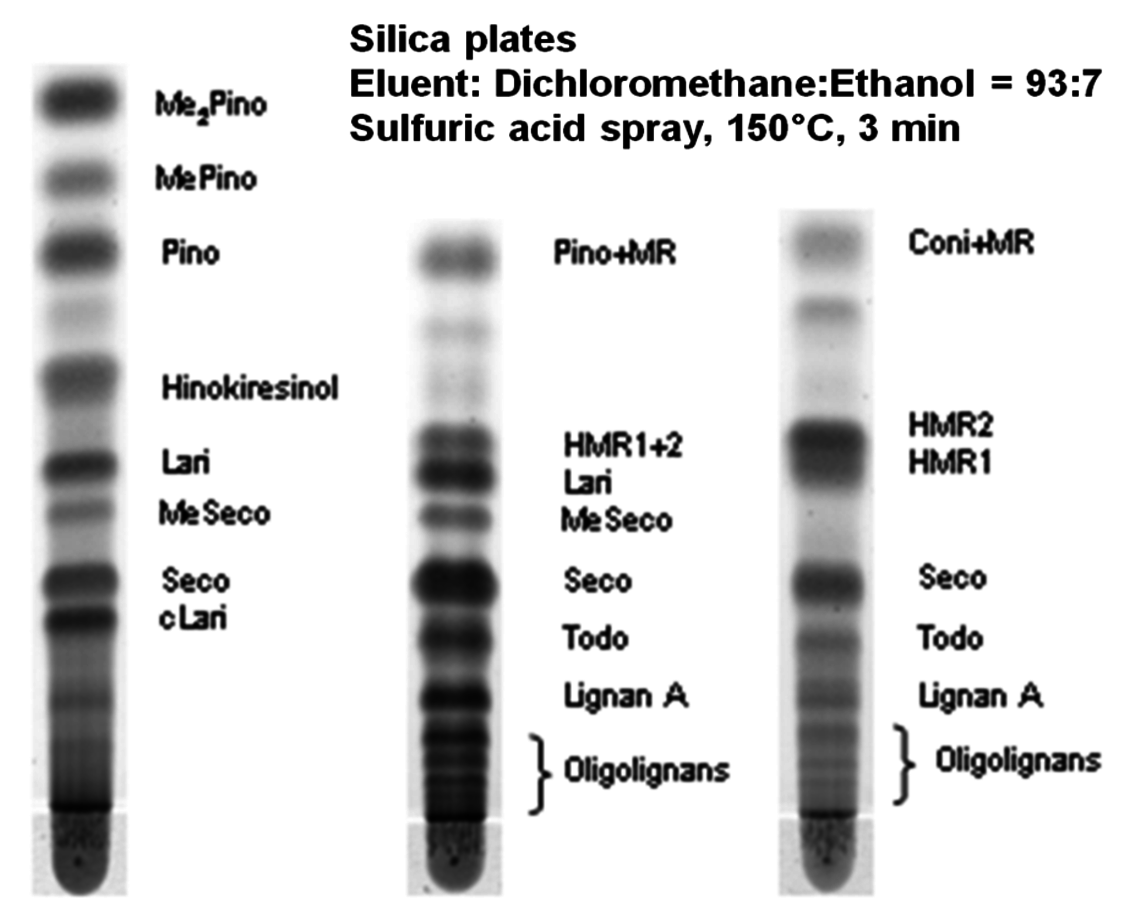
Fig. 7.9: TLC of ethanol extracts of knots from: Araucaria angustifolia (left), Abies alba (center), Picea abies trees (right) [21]
Nevertheless, there is a large potential in the application of LC-MS toward analysis of oligosaccharides, lignans and oligolignans, flavonoids, stilbenes and tannins, and even fragments of lignin [21].
One form of LC, which is still used in organic synthesis and was popular until the 1960s in the analysis of monosaccharides obtained by hydrolysis of wood, is the so-called planar chromatography or thin-layer chromatography (TLC), where the separation is done in paper sheets or on particle layers deposited on glass, plastic or aluminium plates. Although these times of analysis of carbohydrates are long gone, TLC is an excellent technique for small scale preparative separation of fractions to be further analyzed by GC or LC. During analysis an eluent and the analytes rise in the stationary phase due to capillary forces. The analytes are separated according to their affinity to the stationary phase, which is most commonly silica (Figure 7.9).
7.3.4 Spectrometric Methods
Besides chromatography a wide variety of other techniques are available, such as capillary electrophoresis (CE), Infra Red spectrometry (IR), Nuclear Magnetic Resonance (NMR), Raman, Near Infra Red Spectrometry (NIR) and Ultra Violet-Visual Light Spectrometry (UV-Vis). Electrophoresis is a separation technique based on the differential transportation of charged species in an electric field through a conductive medium. Capillary electrophoresis (CE) was designed to separate species depending on their size to charge ratio in the interior of a small capillary filled with an electrolyte and can be used for analyzing oligosaccharide and monosaccharide reaction products. In the current review we focus mainly on chromatographic methods although the spectrometric methods listed above are certainly of great importance. For instance, UV spectrometry can be used for the determination of lignins in solutions. Colorimetric methods based on selective complexation with special reagents, which can be determined by spectrometric measurements in the UV-Vis range, are applied for the determination of metal ions, hemicelluloses and pectins. IR is a possibility to identify such functional groups as hydroxyls, carbonyls, carboxyls and amines.
For example, the analysis of products in rapeseed oil hydrogenation was conducted by IR [22]. However, IR spectra of large biomolecules are complex, moreover spectra of component mixtures could be difficult to interpret. An advantage of Raman spectroscopy for the transformation of biomass occurring often in water solutions is the easy detection of double and triple carbon-carbon bonds while the adsorption of water is weak. Thus, in contrast to FTIR, wet pulp and wood samples can be analyzed with signals related to extractives, lignin and carbon hydrogen bonds of the polysaccharides, while in FTIR signals of the hydroxyl groups of wood polysaccharides are dominating. NMR is an important method as it provides structural information about complex molecules, therefore it is frequently used for structural analysis of lignins and even hemicelluloses. Crystalline cellulose requires the application of solid-state NMR, as utilized for instance recently in the hydrolytic hydrogenation of cellulose [17].
7.3.5 Selection of Analytical Methods
Summarizing shortly the methods described above, it can be stated that the choice of analytical methods in general depends on sample characteristics, matrix complexity, the aim of the analysis, accessible equipment and the amount of resources available.
For instance in order to be analyzed by GC, compounds in the samples must be able to get volatilized and additionally possess thermostability. In case of LC, solubility in the mobile phase is important as well as size, structure and hydrophobicity, presence of functional groups, etc.
Regarding matrix complexity it could be also mentioned that chromatography can be used both for the separation of a compound from the matrix and for quantification and identification. It is important but rarely considered that no residual matter should remain in the samples; especially heavy compounds, which are difficult to evaporate in the GC columns, could significantly influence subsequent analyses. Thus, regular control of retention times and response factors, as well as column cleaning or replacement in due time should not be overlooked. For some samples related to the analysis of biomass even prefractionation could be necessary.
7.4 Analytical Examples
7.4.1 Analysis of Carbohydrates and their Transformation Products
Let us consider first the analysis of a sample of hemicelluloses dissolved in water. The general analytical strategy is given in Figure 7.10. An analytical procedure using GC based on acid methanolysis consists of the following steps [23]. Freeze drying of a 2 mL solution of hemicellulose in water with the subsequent addition of 2 M HCl in water-free methanol, is followed by keeping the sample at 100 °C for three hours of neutralization with pyridine, addition of internal standard (sorbitol), evaporation, silylation (hexamethyldisilazane and trimethylchlorosilane), and finally GC analysis. The latter could use, for instance, a split injector (260 °C, split ratio 1:15) with a 30 m/0.32 mm i.d. column coated with dimethyl polysiloxane (DB-1, HP-1), hydrogen or helium as a carrier gas and FID with a following temperature programme: 100–280 °C and ramping 4 °C/min.
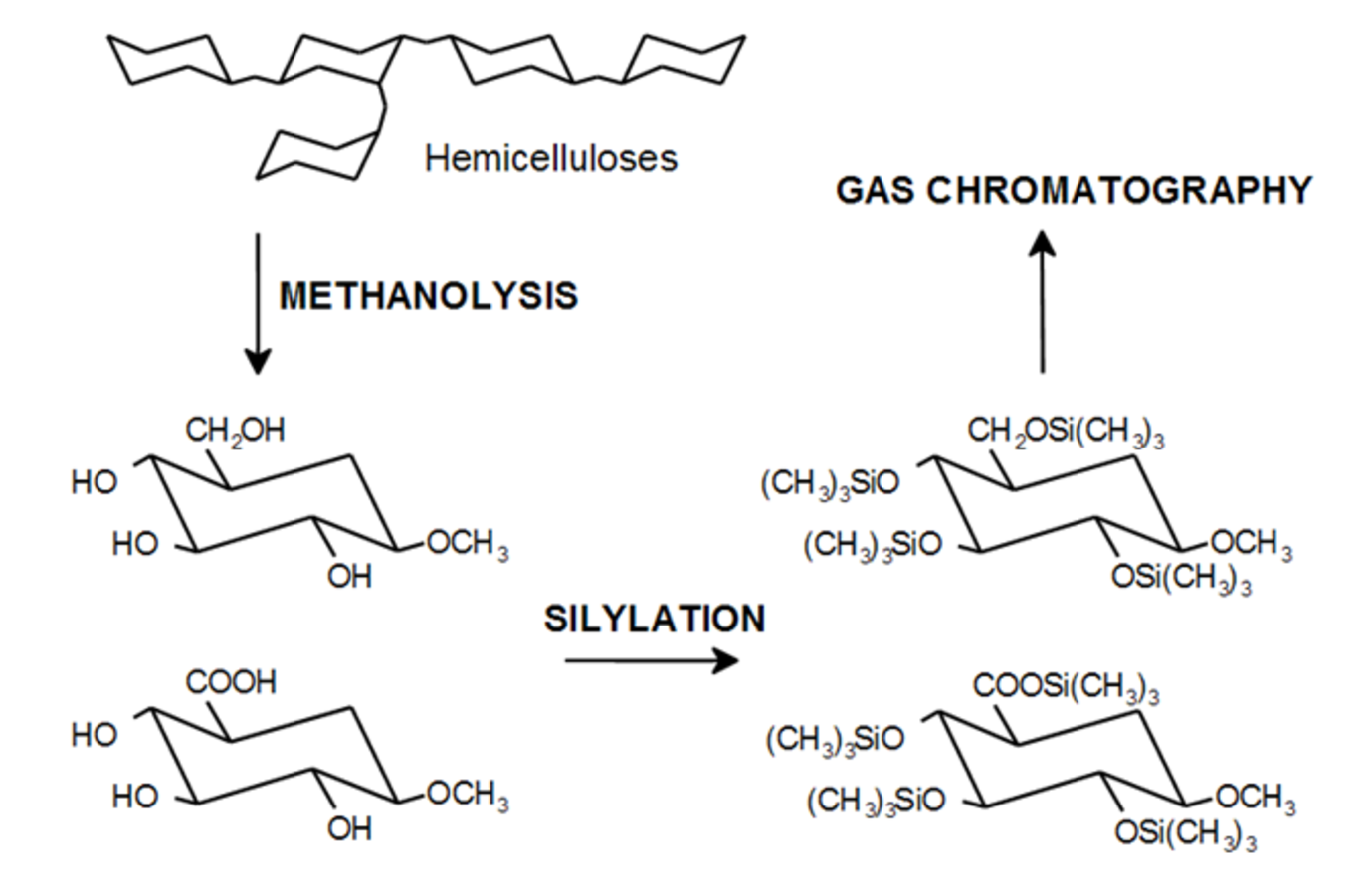
Fig. 7.10: Analysis of sugar units in hemicellulose
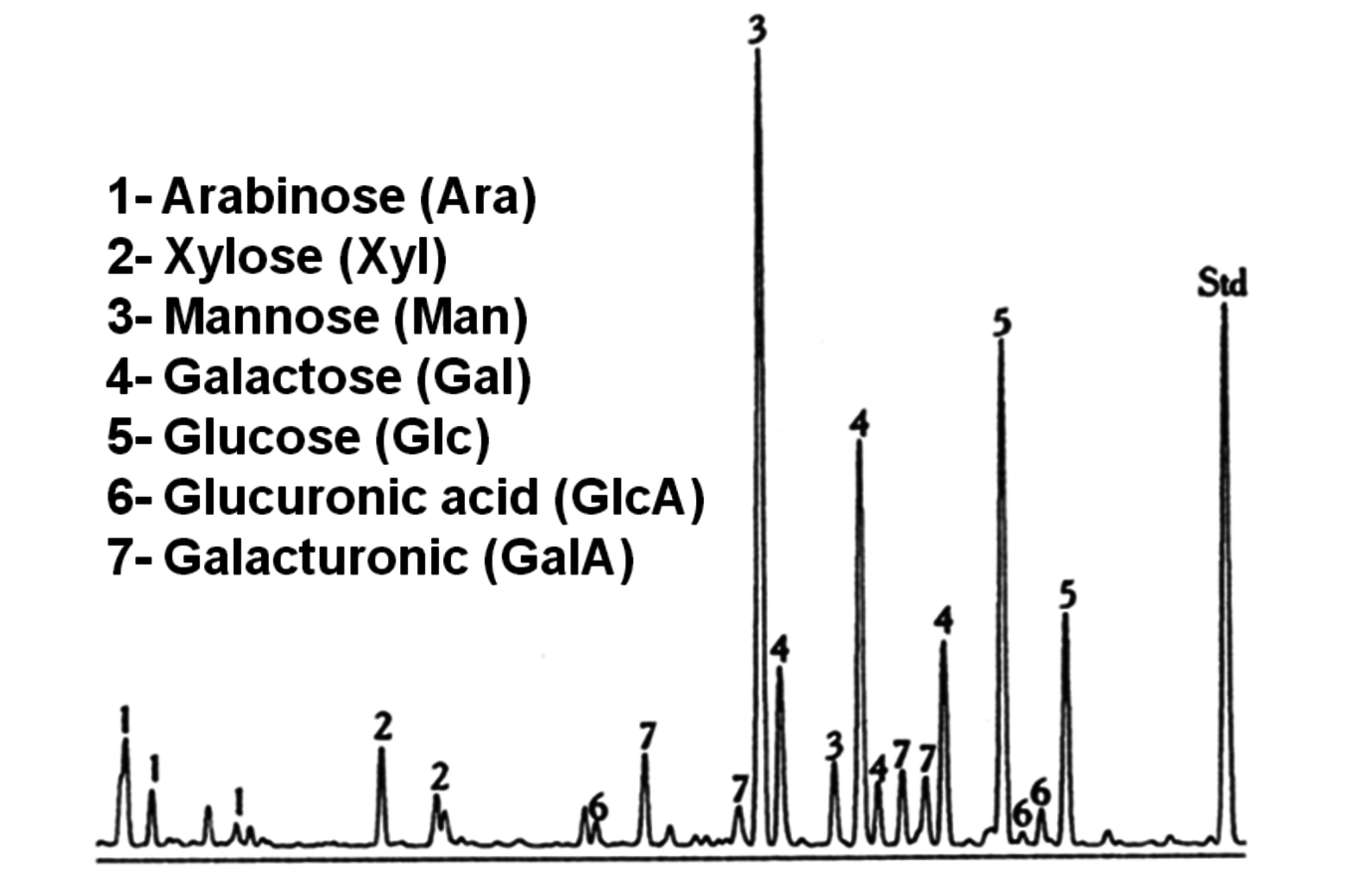
Fig. 7.11: Typical gas chromatogram showing the major sugar units released upon methanolysis of a sample (spruce wood) containing hemicelluloses (Std = internal standard, sorbitol)
An advantage of direct methanolysis of wood samples is that essentially only hemicelluloses are cleaved and very little cellulose. Moreover, contrary to hydrolysis, it allows less degradation of released monosaccharides. Methanolysis can be used also for direct analysis of solid wood and fiber samples. A typical chromatogram is presented in Figure 7.11, showing several peaks for a particular sugar due to the presence of α & β anomers of pyranoses & furanoses (Figure 7.12).
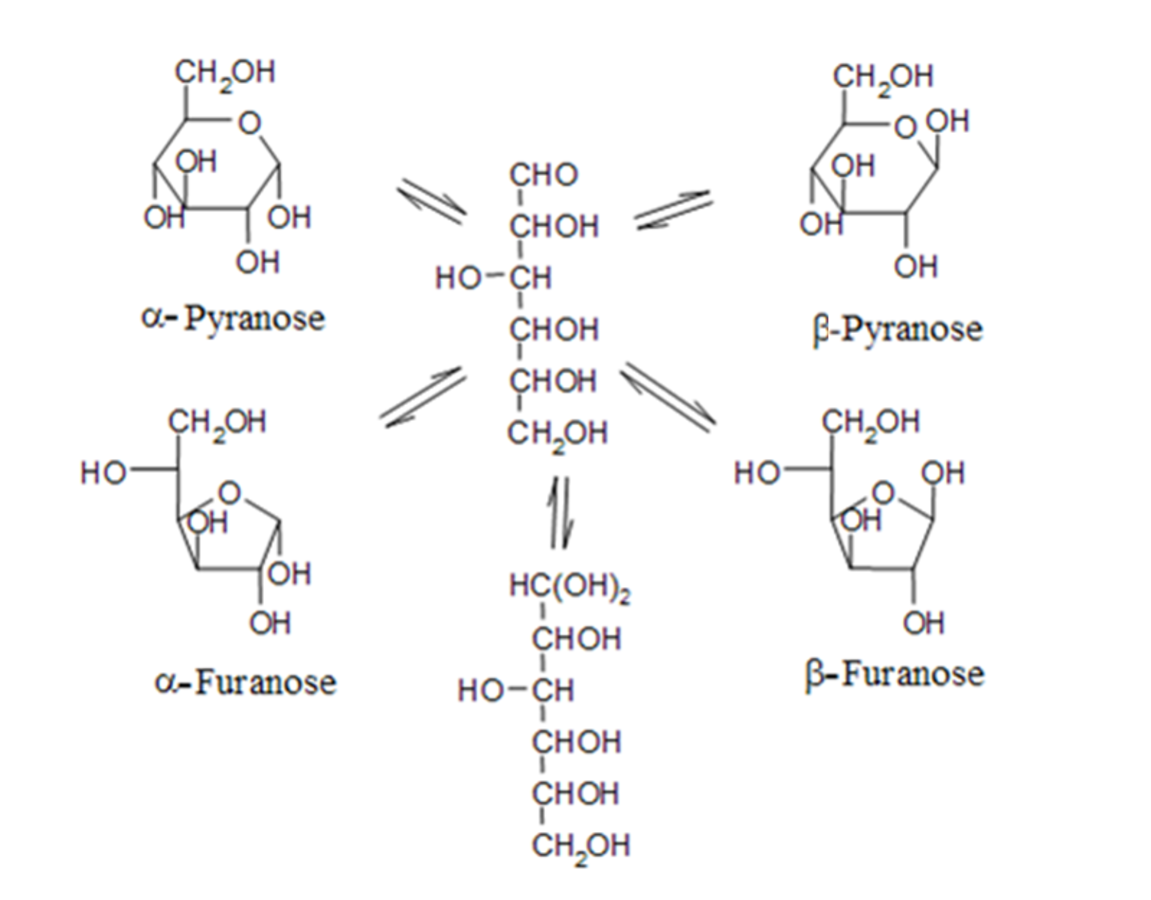
Fig. 7.12: Equilibrium of different forms of sugars
Due to the complexity of the product mixture and the analytical procedure correction factors are needed. For instance, cleavage (the methanolysis) could be incomplete for certain glycosidic bonds, such as the Xyl-MeGlcA bond. Some degradation of formed sugars, especially uronic acids may happen and the products can have different detector responses. In order to determine correction factors it is recommended to perform methanolysis, silylation and GC analysis on a sample containing equal amounts of Ara, Xyl, Man, Glc, Gal, GlcA, GalA, etc., and pure hemicelluloses and pectins (if present) and to compare peak areas with the area of the internal standard.
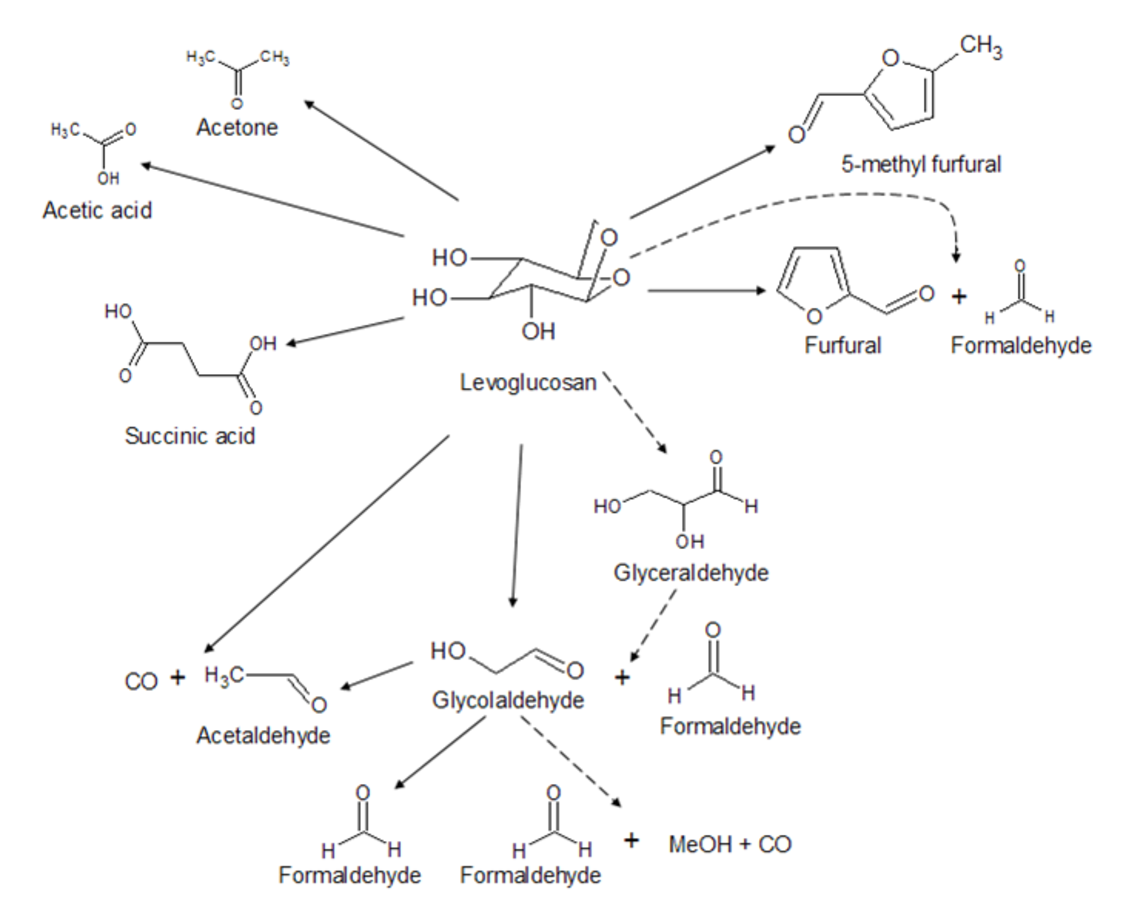
Another example worth considering is the gas-phase catalytic transformation of levoglucosan over zeolites [24,25]. The reaction scheme is given in Figure 7.13. In [24,25] for HPLC analysis an acid Aminex cation H+ column with sulfuric acid (0.005 M) as a mobile phase with a flow of 0.5 ml/min at 338 K was used, along with an Aminex HPX-87C column and mobile phase-calcium sulfate (1.2 mM) with a flow rate of 0.4 ml/min at 353 K. A refractive index detector was applied. Figure 7.14 illustrates that the separation is very much dependent on the analytical conditions.
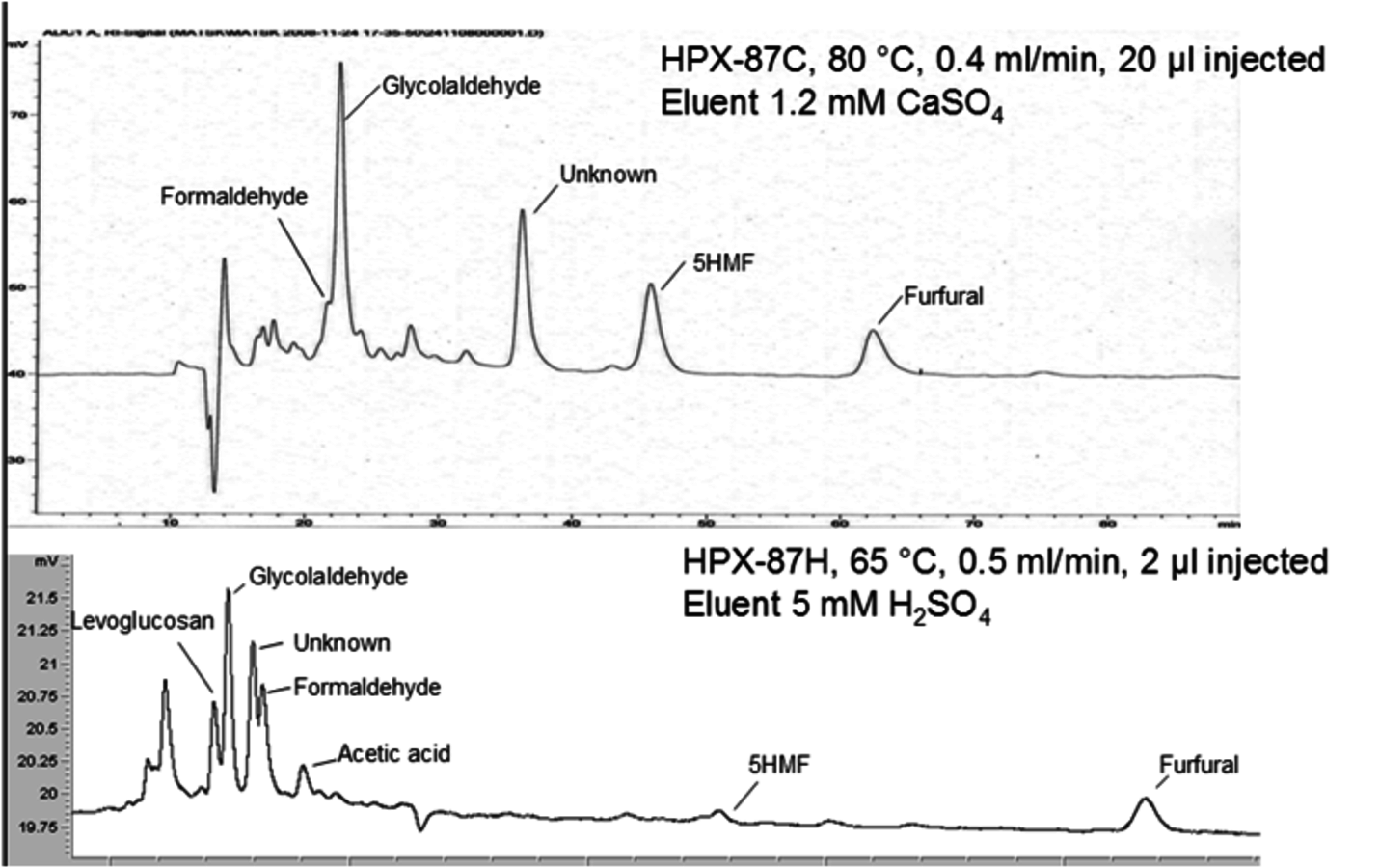
Fig. 7.14: Analysis of a levoglucosan transformation mixture by HPLC with two different columns [26]
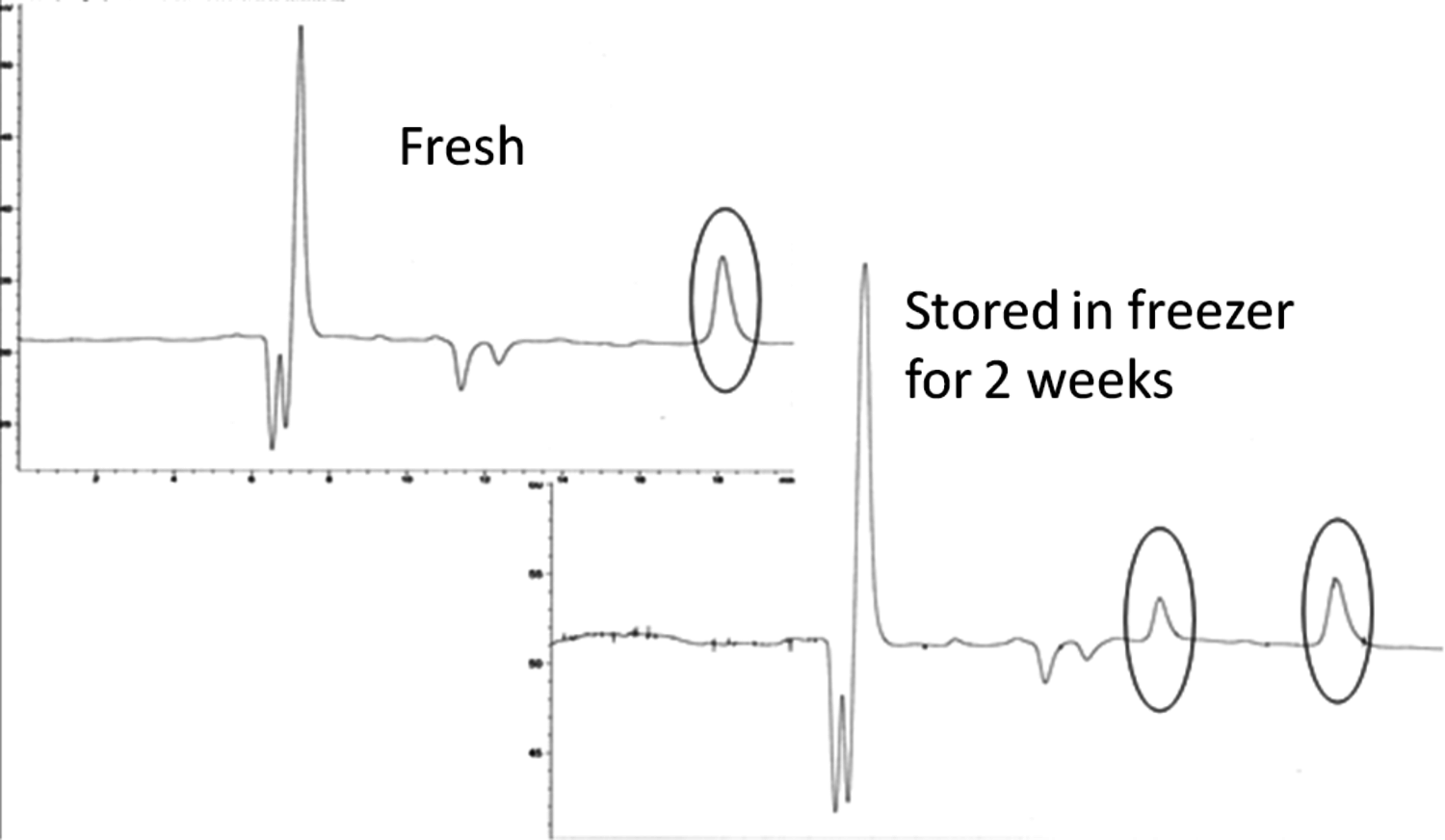
Fig. 7.15: HPLC data showing instability of reaction products in levoglucosan transformations [26]
Stability of the samples is another important issue, which should also be carefully considered, as illustrated in Figure 7.15. Samples stored in a freezer exhibited another peak, which is certainly a result of transformations happening during storage.
An even more prominent difference in analysis was noticed in the aqueous reforming of sorbitol [27-29]. Comparison of the analysis for different columns is given in Figure 7.16 demonstrating that for the identification of reaction products tedious and time-consuming analytical work is required.
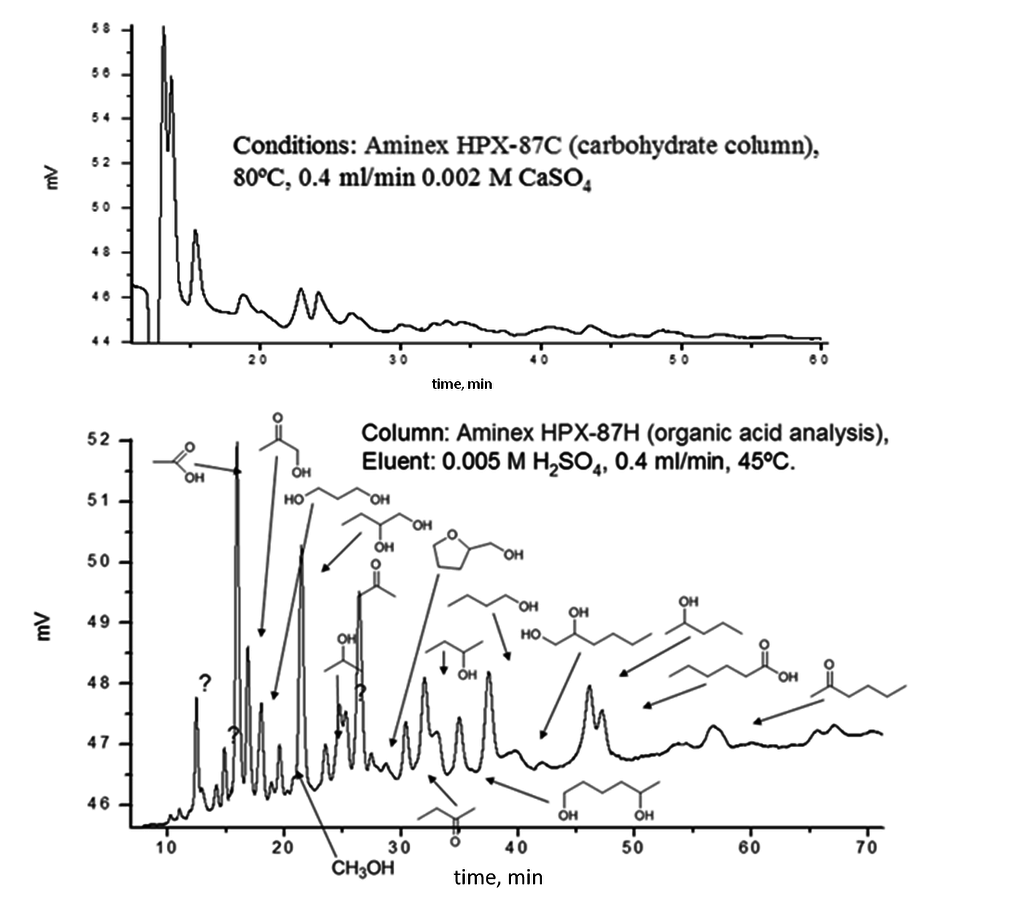
Fig. 7.16: HPLC analysis of aqueous phase reforming products [29]
7.4.2 Analysis of Lignin
Structural information on lignins could be obtained by wet-chemical and spectroscopic methods using the approach for analysis of wood given in Figure 7.17.
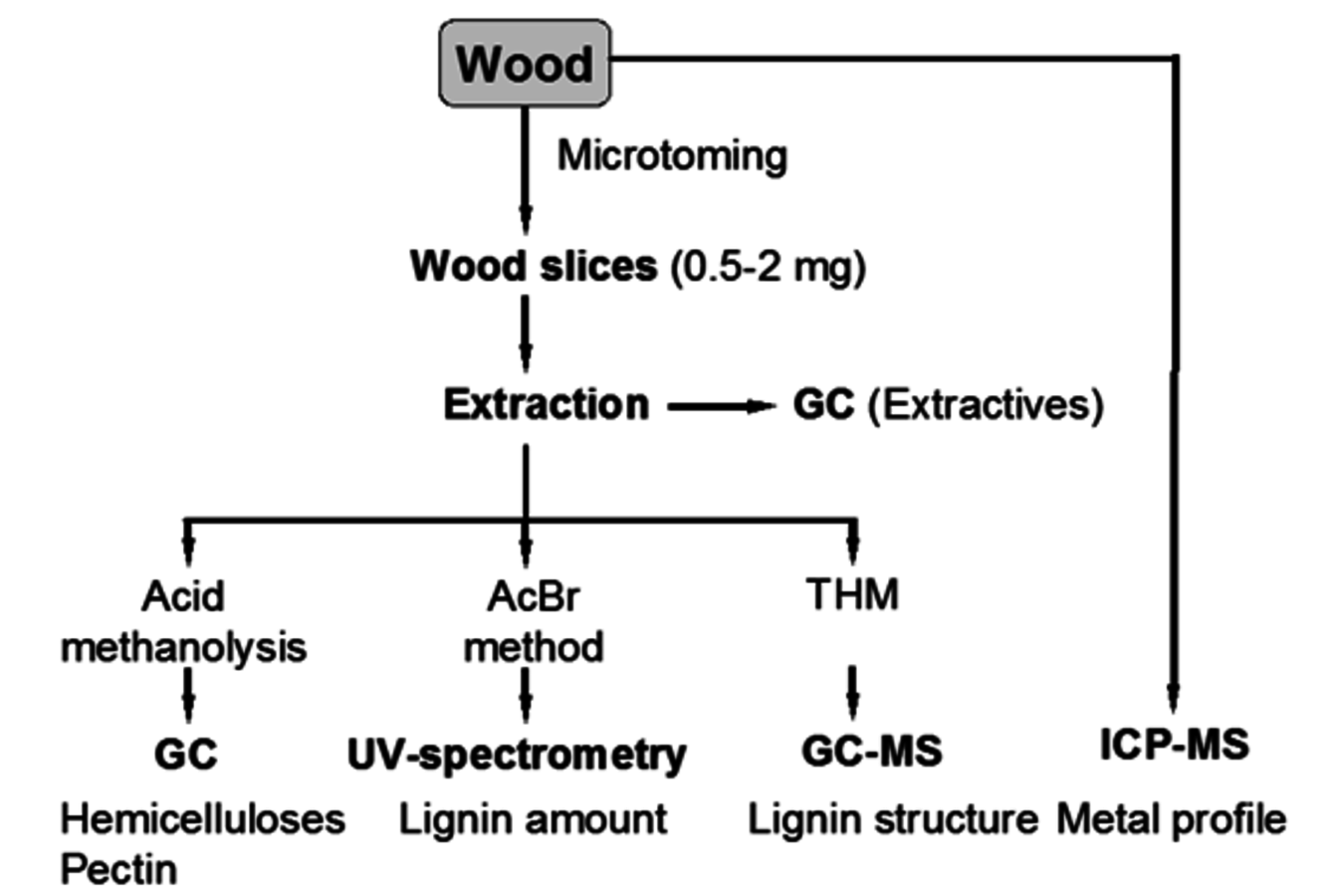
Fig. 7.17: A scheme for the microanalysis of wood [30]
Here, 5 mL 20% AcBr in pure acetic acid is added to ca. 1–10 mg of wood followed by the addition of 0.1 mL perchloric acid (70%) and keeping the mixture for 3 hours at 50 °C, with subsequent neutralization with NaOH, dilution and UV-Vis analysis at 280 nm.
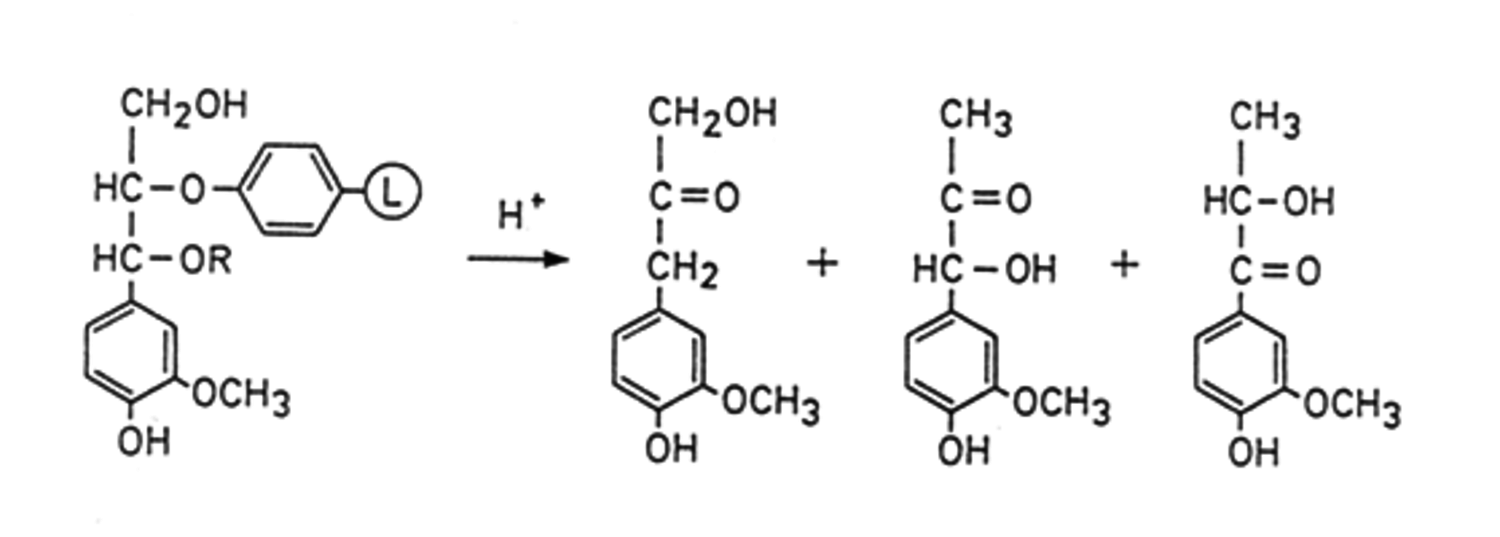
Fig. 7.18: Acidolysis of lignin
Direct analysis of lignin in wood can be performed by selective / specific degradation followed by GC analysis. Among degradation methods acidolysis, thioacidolysis, permanganate oxidation and pyrolysis can be mentioned. Acidolysis (Figure 7.18) cleaves predominantly β-O-4-ether bonds by acid hydrolysis and gives many degradation products with a rather low yield of ca. 60%.
Thioacidolysis (Figure 7.19) gives selective cleavage of β-O-4-ether bonds and results in less complex mixtures than acidolysis and also gives higher yields (> 80%) being able to quantify units with β-O-4-ether bonds and a free hydroxyl. This reaction is performed in dioxane-ethanethiol with boron trifluoride etherate. The degradation products are silylated prior to analysis by GC.
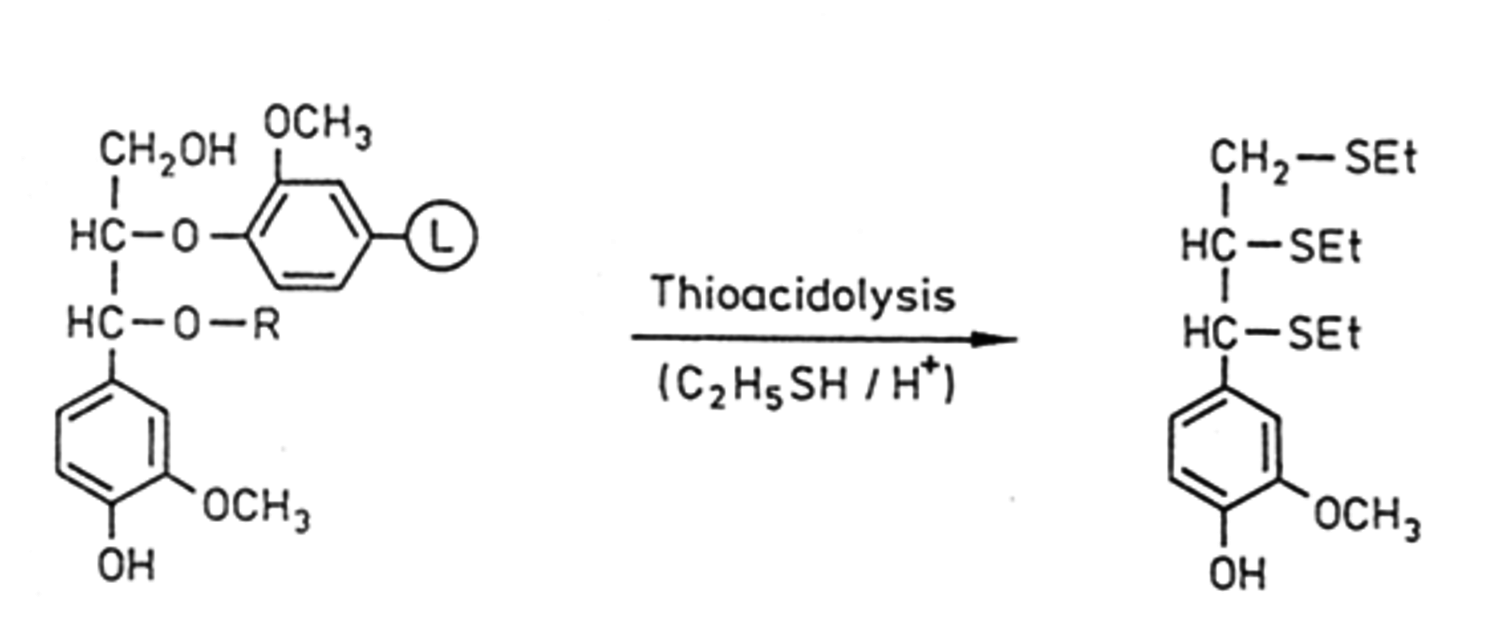
Fig. 7.19: Thioacidolysis of lignin
Oxidation by permanganate (KMnO4 – NaIO4 at 82 °C for 6 hours) in acidic solution of ethylated (at pH = 11 and 25 °C with diazoethane for 25 hours) samples with further oxidation by alkaline peroxide for 10 min at 50 °C and final methylation results in samples which can be analyzed by GC (Figure 7.20). This method gives information only on aromatic units with a free hydroxyl, comprising about 10% of lignin in wood and ca. 70% of lignin after kraft cooking.
Another method for the analysis of lignin is pyrolysis combined with GC-MS (Py GC-MS) allowing even a simultaneous determination of lignin and carbohydrates. Py GC-MS can be combined with advanced chemometric methods such as principal component analysis to enable a more complete identification of various lignin fragments. In summary, it can be stated that because of the heterogeneity of lignin there is no universal degradation method giving all desired information on the lignin structure, however, by combination of several methods the structure of lignin can be described fairly well.
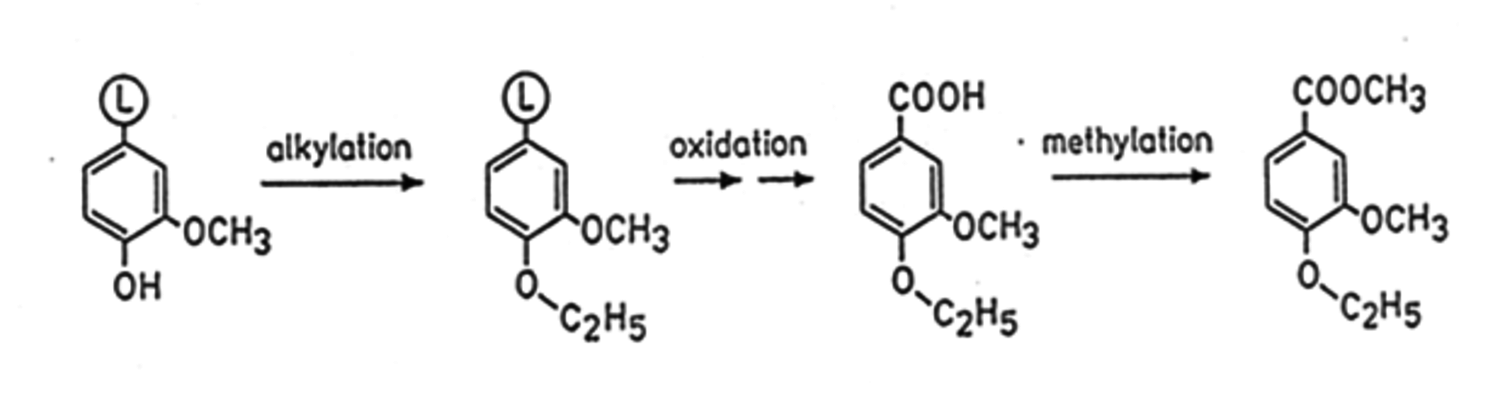
Fig. 7.20: Oxidation method for the analysis of terminal units (free phenolic groups) in lignin
7.4.3 Wood Extractives
Wood contains a wide variety of components that are extractable with various organic solvents or water. Non-polar and semi-polar solvents (hexane, dichlorome-
thane, diethyl ether, MTBE etc.) extract lipophilic oleoresin and fat components, while polar solvents (acetone, ethanol, water, etc.) extract hydrophilic phenolics, sugars, starch and inorganic salts. Acetone and ethanol extract also lipophilic extractives.

Fig. 7.21: Classification of wood extractives [31]
A classification of wood extractives is given in Figure 7.21, while the analytical procedure for extractives is outlined in Figure 7.22. Group analysis of fatty acids, resin acids, triglycerides, lignans and sterols can be done using a short column GC (5–7 m/0.53 mm capillary column with 0.15 μm film thickness), or by HP-SEC (Figure 7.23) as well as thin layer chromatography.
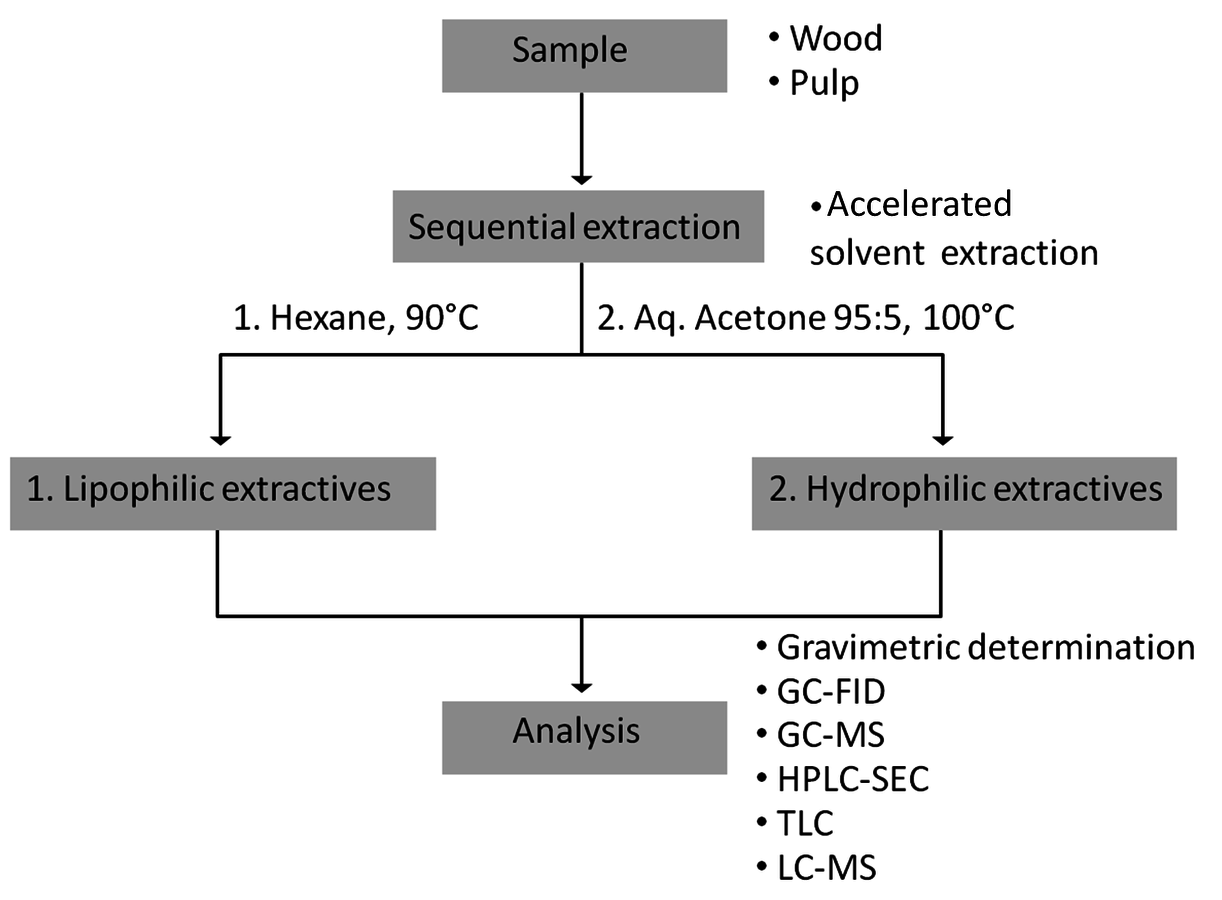
Fig. 7.22: Analytical procedures for wood extractives
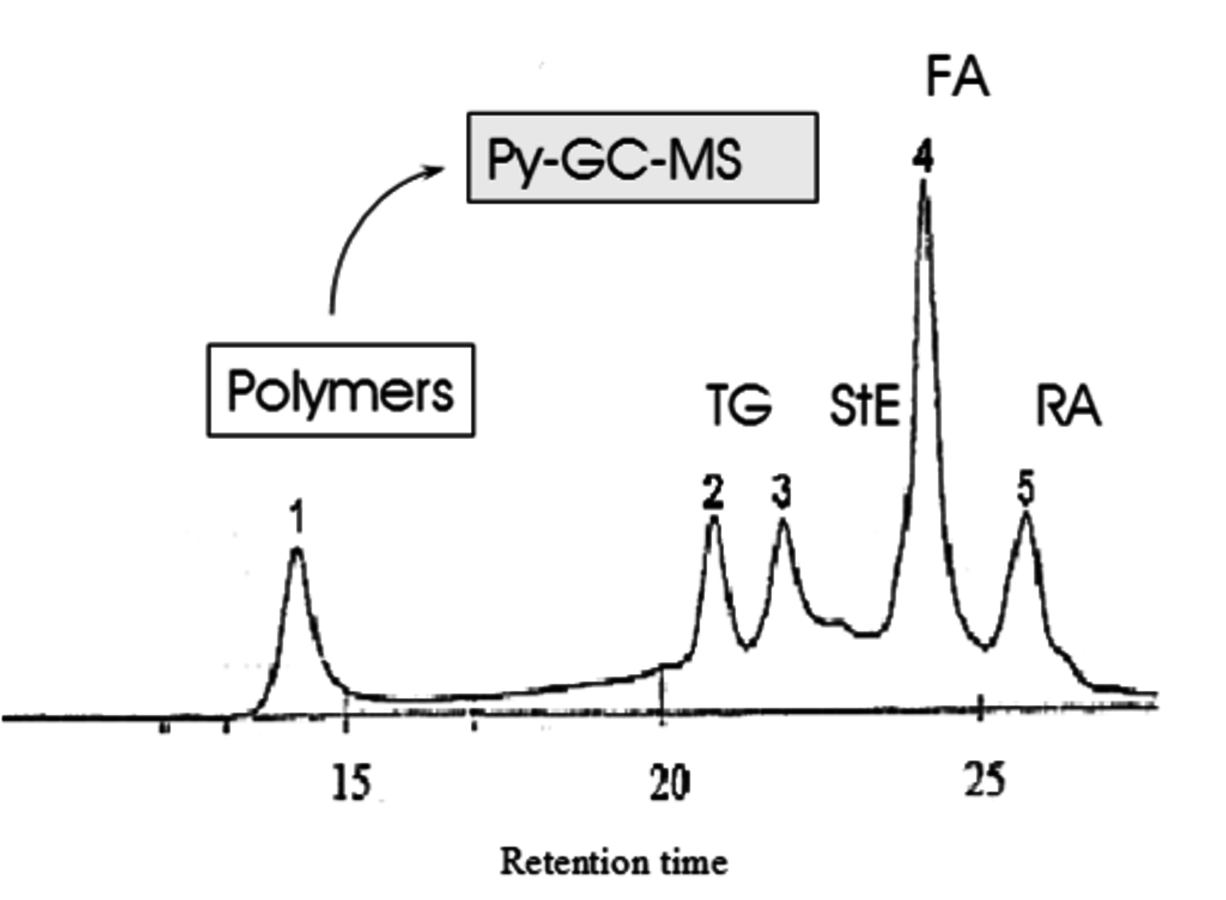
Fig. 7.23: Separation of wood extractives with SEC. TG, StE, FA and RA stand for triglycerides, sterols, fatty acids and resin acids respectively [32].
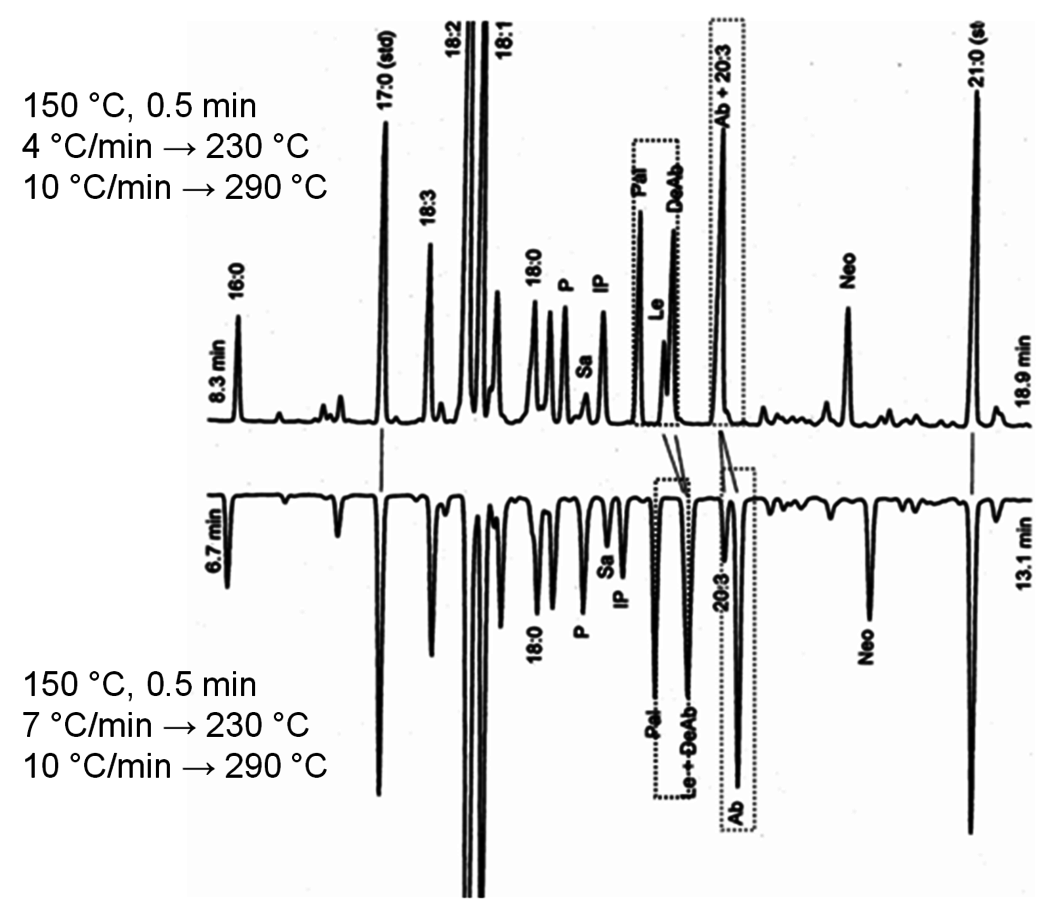
Fig. 7.24: GC of fatty acids and resin acids with HP-1, 30 m, 0.32 mm i.d. column with different temperature gradients [31]
The analysis of individual compounds can be done by GC on a longer column (20–30 m/0.20–0.32 mm capillary columns) and reverse phase HPLC, while the identification of compounds can be performed by GC-MS, LC-MS, and NMR of isolated substrates. In case of a poor separation between compounds, the following parameters could be modified: temperature gradient, column polarity, type of derivative used in derivatization. As seen in Figure 7.24 a better separation, for example, between abietic acid and tri-unsaturated C20 fatty acid, is achieved with somewhat higher ramping. The HP-1 column usually follows a boiling point order, however, columns with different polarity could also be used (Figure 7.25) to allow better separation.
As previously mentioned, derivatization of fatty and resin acids is needed for accurate quantitative analysis. Although methylation is a commonly used method, silylation can sometimes afford better separation (Figure 7.26). In addition, for some GC columns peak-tailing is more severe for methyl esters than for silyl esters.
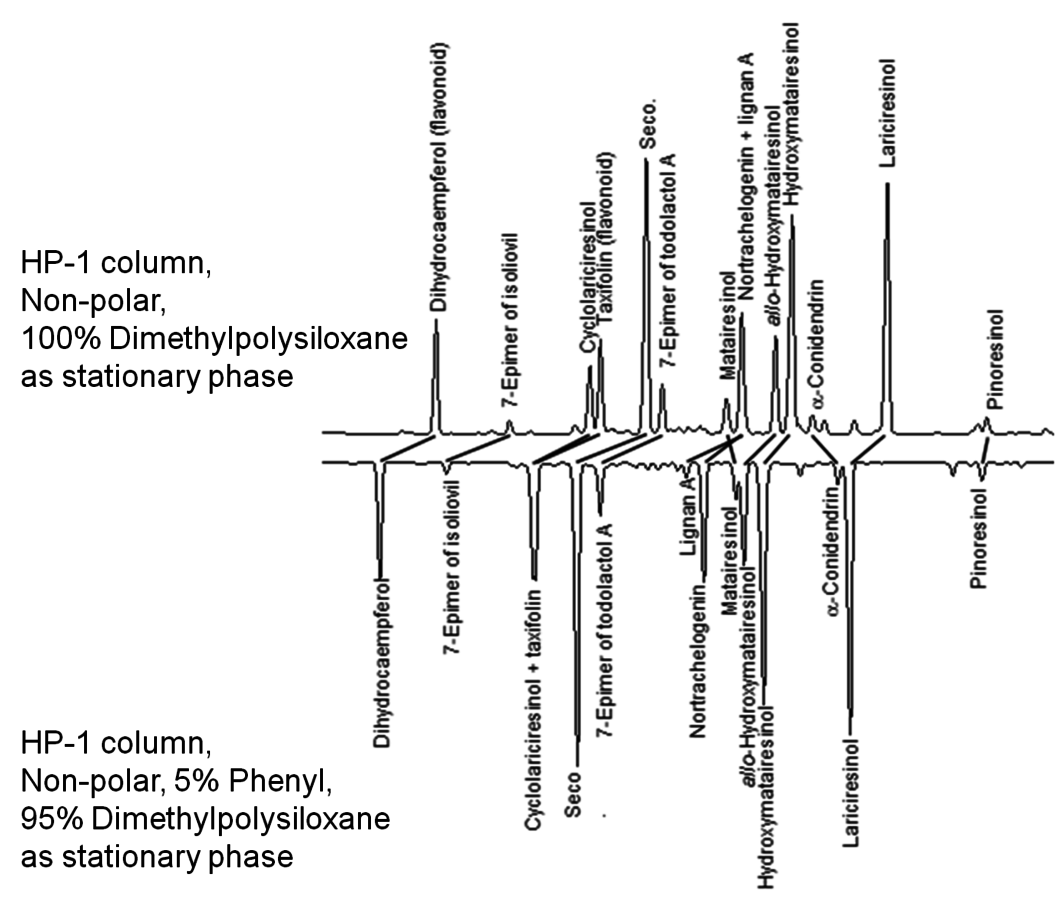
Fig. 7.25: GC separation of phenolic extractives (flavonoids and lignans) using columns of different polarity [21]
7.5 Final Words
This chapter describes some of the contemporary methods for the chemical analysis of biomass-derived chemicals. All available methods could not have been treated in this review, therefore the focus was mainly on chromatographic methods. A more comprehensive overview of analytical methods was published several years ago by one of the authors [31,33]. In the current work, detailed procedures were discussed for only a few cases as the emphasis was laid more on general approaches.
The analytical procedure depends very much on the objective of a particular study as well as the available resources in terms of instruments, time, costs and human skills.
The main hurdles on the way toward the development of a reliable analytical method for a particular application are associated with a lack of time to check methods described in literature, a certain trust in already published procedures, even if they are far from being perfect, as well as a pressure from granting agencies/sponsors to get “real” catalytic results rather than means to develop or check analytical methods. In the latter case there is certainly more glory in developing new methods compared to just checking the old ones.
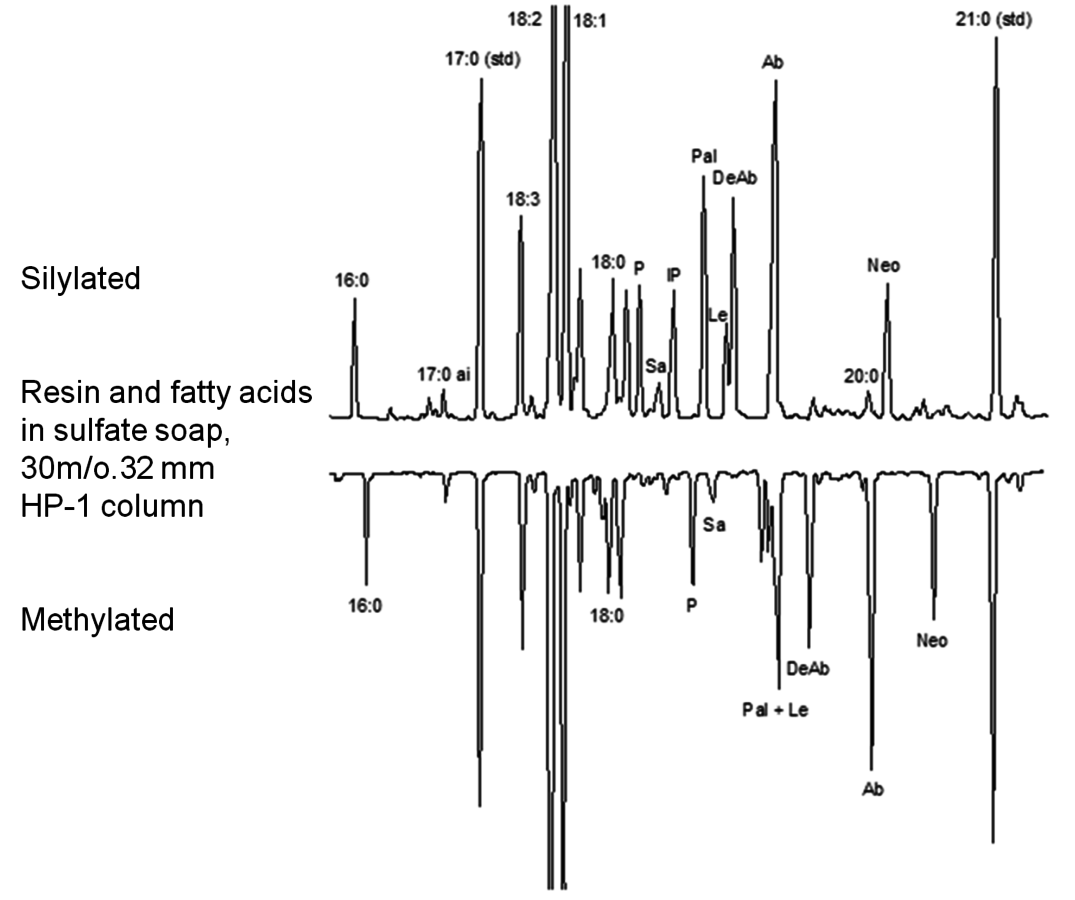
Fig. 7.26: Comparison between GC analysis of methylated and silylated fatty and resin acids [31]
Finally, we should stress that no single method works perfectly for all kinds of samples. Moreover, dubious methods are sometimes presented in literature, which means that the results are not reliable. It can thus be emphasized once more that improving analytical methods, not only in the particular case of the catalytic transformation of biomass, but in general improves the quality of science.
Bibliography
[1] C. Okkerse, H. Bekkum. From Fossil to Green Green Chemistry 4: 107-114 (1999): 107-114.
[2] P.T. Anastas, J.C. Wagner. Green Chemistry Theory and Practice. New York: Oxford University Press, 1998
[3] T. Werpy, G. Petersen. Top Value Added Chemicals from Biomass, Vol 1: Results of Screening for Potential Candidates from Sugars and Synthesis Gas. Battelle: US Department of Energy, Energy EfficiencyRenewable Energy,, 2004
[4] G.W. Huber, S. Iborra, S. I.. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering Chemical Reviews 106: 4044-4098 (2006): 4044-4098.
[5] L. Petrus, M.A. Noordermeer. Biomass to Biofuels, a Chemical Perspective Green Chemistry 8: 861-867 (2006): 861-867.
[6] G.W. Huber, A. Corma. Synergies between Bio- and Oil Refineries for the Production of Fuels from Biomass Angewandte Chemie International Edition (2007):.
[7] S. Lestari, P. Mäki-Arvela, P. MA., Beltramini P., B. P.. Transforming Triglycerides and Fatty Acids to Biofuels ChemSusChem 2: 1109-1119 (2009): 1109-1119.
[8] R. Rinaldi, F. Schüth. Design of Solid Catalysts for the Conversion of Biomass Energy & Environmental Science 2: 610-626 (2009): 610-626.
[9] D.M. Alonso, J.Q. Bond, J. B., Serrano-Ruiz J.Q.. Catalytic Conversion of Biomass to Biofuels Green Chemistry 12: 1493 (2010): 1493.
[10] J. Zakzeski, P.C.A. Bruijnincx, P. B., Jongerius P.C.A.. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals Chemical Reviews 110: 3552-3599 (2010): 3552-3599.
[11] E. Adler. Lignin Chemistry - Past, Present and Future Wood Science and Technology 11: 169-218 (1977): 169-218.
[12] A. Atutxa, R. Aguado, R. A., Gayubo R., G. R.. Kinetic Description of the Catalytic Pyrolysis of Biomass in a Conical Spouted Bed Reactor Energy Fuels 19: 765-774 (2005): 765-774.
[13] P. Mäki-Arvela, B. Holmbom, B. H., Salmi B.. Recent Progress in Synthesis of Fine and Specialty Chemicals from Wood and Other Bio- mass by Heterogeneous Catalytic Processes Catalysis Reviews, Science and Engineering 49: 197 (2007): 197.
[14] A. Fukuoka, P.L. Dhepe. Catalytic Conversion of Cellulose into Sugar Alcohols Angewandte Chemie International Edition 45: 5161-5163 (2006): 5161-5163.
[15] P.L. Dhepe, A. Fukuoka. Cellulose Conversion under Heterogeneous Catalysis ChemSusChem 1: 969-975 (2008): 969-975.
[16] A. Fukuoka, P.L. Dhepe. Sustainable Green Catalysis by Supported Metal Nanoparticles The Chemical Record 9: 224-235 (2009): 224-235.
[17] V. Jolle, F. Chambon, F. C., Rataboul F., R. F., F. Rataboul, F. R.. Non-Catalyzed and Pt/Gamma-Al Green Chemistry 11: 2052-2060 (2009): 2052-2060.
[18] F. Oersa, B. Holmbom. A Convenient Method for the Determination of Wood Extractives in Papermaking Process Waters and Effluents Journal of Pulp and Paper Science 20: J361-J365 (1994): J361-J365.
[19] S. Willför, A. Pranovich, A. P., Tamminen A., T. A., T. Tamminen, T. T., Puls T., P. T., J. Puls, J. P.. Carbohydrate Analysis of Plant Materials with Uronic Acid-Containing Polysaccharides - A Comparison between Different Hydrolysis and Subsequent Chromatographic Analytical Techniques Industrial Crops and Products 29: 571-580 (2009): 571-580.
[20] P. Tolvanen, P. Mäki-Arvela, P. MA., Kumar P., K. P., N. Kumar, N. K., Eraenen N., E. N.. Thermal and Catalytic Oligomerisation of Fatty Acids Applied Catalysis A: General 330: 1-11 (2007): 1-11.
[21] S.M. Willför, A.I. Smeds, A. S.. Chromatographic Analysis of Lignans Journal of Chromatography A 1122: 64-77 (2006): 64-77.
[22] I.V. Deliy, N.V. Maksimchuk, N. M., Psaro N.V., P. N., R. Psaro, R. P., Ravasio R., R. R.. Kinetic Peculiarities of cis/trans Methyl Oleate Formation during Hydrogenation of Methyl Linoleate over Pd/MgO Applied Catalysis A: General 279: 99-107 (2005): 99-107.
[23] A. Sundberg, K. Sundberg, K. S., Lillandt K.. Determination of Hemicelluloses and Pectin in Wood and Pulp Fibres by Acid Methanolysis and Gas Chromatography Nordic Pulp & Paper Research Journal 226: 216-219 (1996): 216-219.
[24] M. Käldström, N. Kumar, N. K., Heikkilä N., H. N., T. Heikkilä. Formation of Furfural in Catalytic Transformation of Levoglucosan over Mesoporous Materials ChemCatChem 2: 539-546 (2010): 539-546.
[25] M. Käldström, N. Kumar, N. K., Salmi N.. Levoglucosan Transformation over Aluminosilicates Cellulose Chemistry and Technology 44: 203-209 (2010): 203-209.
[26] M. Käldström, D.Yu. Murzin. unpublished data. .
[27] A.V. Kirilin, A.V. Tokarev, A. T., Murzina A.V., M. A., E.V. Murzina. Reaction Products and Intermediates and their Transformation in the Aqueous Phase Reforming of Sorbitol ChemSusChem 3: 708-718 (2010): 708-718.
[28] A.V. Tokarev, A.V. Kirilin, A. K.. unpublished data. .
[29] A.V. Tokarev, A.V. Kirilin, A. K., Murzina A.V., M. A., E.V. Murzina, E. M.. The Role of Bioethanol in Aqueous Phase Reforming to Sustainable Hydrogen International Journal of Hydrogen Energy 35: 12642-12649 (2010): 12642-12649.
[30] A. Pranovich, J. Konn, J. K.. Methodology for Chemical Microanalysis of Wood. In: Proceedings 9th European Workshop on Lignocellulosics and Pulp . 2006. 436-439.
[31] B. Holmbom. Extractives. In: Analytical Methods on Wood Chemistry, Pulping and Papermaking Berlin: Springer Verlag, 1998. 125-148.
[32] B. Holmbom. unpublished data. .
[33] B. Holmbom, P. Stenius. Papermaking Science and Technology. In: Forest Products Chemistry , 2000. 105-169.