13.1 Introduction
The supramolecular structure of cellulose is perhaps the most difficult hurdle facing processes starting from cellulosic fibers. Nonetheless, the supramolecular structure can be disassembled, for example, by dissolving the biopolymer. As a result, cellulose, a recalcitrant polymer in solid-state, becomes a reactive macromolecule in solution. For instance, the hydrolysis of cellulose can proceed even at room temperature in solution; however, the reaction sounds inapplicable below 180 °C when starting from cellulose slurries in water. Although processing cellulose in solution could well hold the key to its efficient conversion into biofuels and bio-based chemical assets, the use of solvents may carry considerable costs. The economics surrounding the use of solvents is beyond the scope of this chapter. Herein, the fundamental challenges facing the hydrolysis of cellulose in solution are discussed. This chapter aims to aid in the chemical understanding of
1cellulose recalcitrance,
2cellulose in solution,
3homogeneous hydrolysis of cellulose, followed by
4final remarks.
13.2 Understanding Cellulosic Recalcitrance
Cellulose is a special polymer. Although it is made of sugar, it is not soluble in water. Moreover, the biopolymer is much less reactive than glucose, its building unit. What makes cellulose so unique? The answer to this question does not lie solely in the structural aspects of a single polymeric chain, but depends also on the supramolecular structure of cellulose, i.e., the chemical system made up of several cellulosic chains commonly called cellulosic microfibril.
Cellulose is a linear polymer, which can be defined either as a syndiotactic polymer of β-D-glucose or as an isotactic polymer of cellobiose [1]. However, defining cellulose as a syndiotactic polymer of β-D-glucose is more common. Thus, the formula of cellulose is
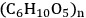 , where n is the number of repeating units or degree of polymerization (DP). Cellulose can exhibit DP as high as 10,000 anhydroglucose units (AGU) [1]. The AGUs are bonded via 1,4-β-glycosidic linkages. This key feature distinguishes cellulose from amylose, another polymer of glucose linked by 1,4-α-glycosidic bonds. The different stereochemistry of the glycosidic linkages leads to a straight chain structure in cellulose and a helical structure in amylose (Figure 13.1) [2].
, where n is the number of repeating units or degree of polymerization (DP). Cellulose can exhibit DP as high as 10,000 anhydroglucose units (AGU) [1]. The AGUs are bonded via 1,4-β-glycosidic linkages. This key feature distinguishes cellulose from amylose, another polymer of glucose linked by 1,4-α-glycosidic bonds. The different stereochemistry of the glycosidic linkages leads to a straight chain structure in cellulose and a helical structure in amylose (Figure 13.1) [2].
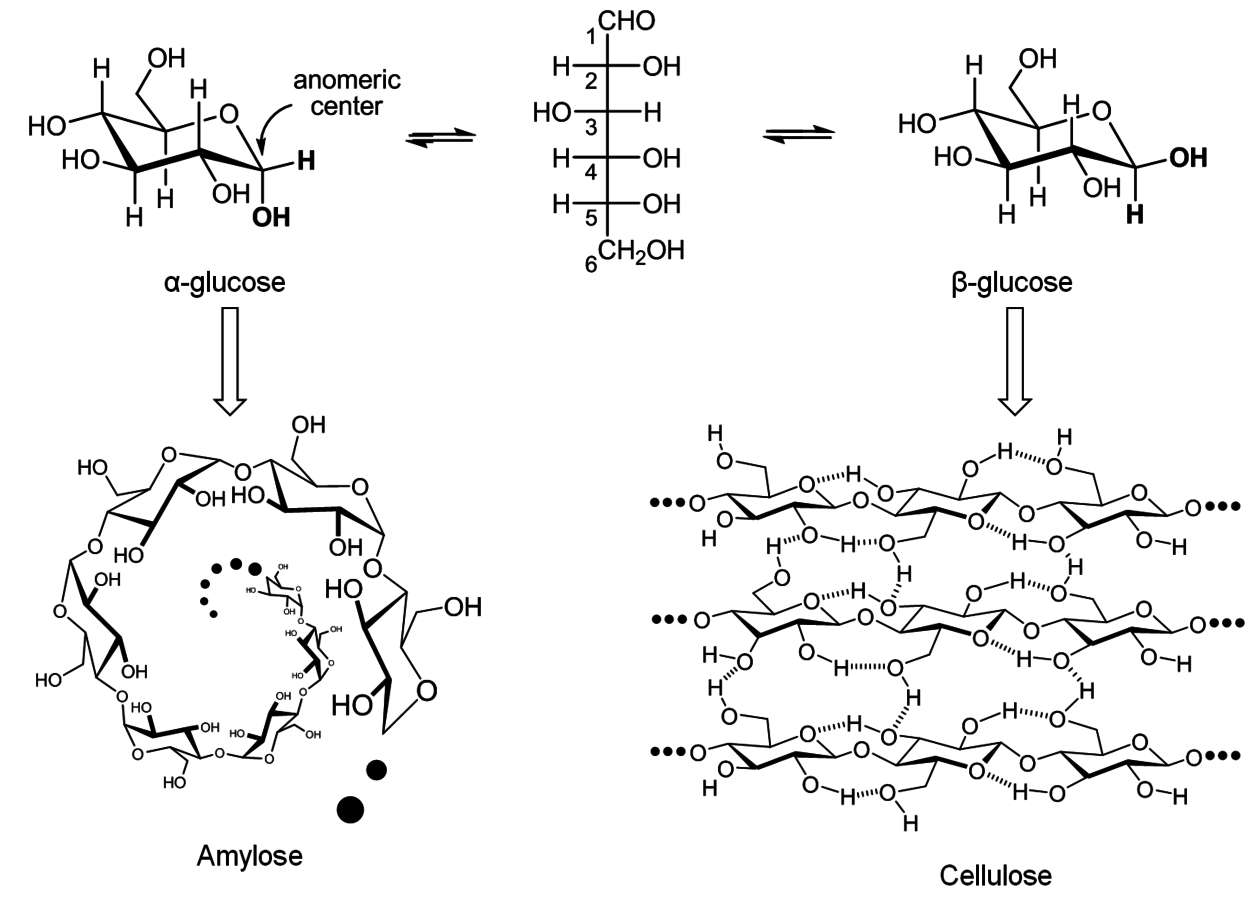
Fig. 13.1: Amylose and cellulose, the polymers of glucose [2]
The 1,4-β-glycosidic linkage enables an intense intramolecular H-bonding among the groups around the glycosidic bond (Figure 13.1) [1]. This makes the polymeric chains assume a straight conformation, which allows them to be packed side-by-side through intermolecular H-bonding [1]. As a result, a planar sheet composed of cellulosic chains is formed. In the polymorph of cellulose found in nature, cellulose I,1 these planar sheets are stacked on one another and held together by van der Waals forces [5]. This supramolecular structure forms the crystalline domain of the microfibrils. In this chemical system, only the cellulosic chains exposed on the surface of the microfibril are easily accessible to solvents, reactants and enzymes (Figure 13.22) [5]. For this reason, the reactivity of cellulose toward hydrolysis is markedly low. Indeed, the half-life of cellulose in water at 25 °C (non-catalyzed hydrolysis) is estimated at 5–8 million years [6].

Fig. 13.2: A computational model of a cellulose microfibril in aqueous solution. At the top, a complete system showing an amorphous region (in the center of the top image) is presented. At lower left, a cross section of the fibril is shown. At lower right, a close-up of the amorphous region is given. Courtesy of Xiaolin Cheng, Oak Ridge National Laboratory
Along the same microfibril both crystalline and amorphous domains are present [3,4]. Considering the catalytic hydrolysis, the reaction proceeds with ease in the amorphous domains, where the structural restrictions are to some extent relaxed [7]. This results in a rapid rate of hydrolysis at the beginning of the reaction. Upon hydrolyzing the amorphous domains, however, the reaction rate progressively slows down reaching negligible rates when only the crystalline domains remain in the suspension [5].
To improve the performance of hydrolytic processes, the supramolecular structure of cellulose should be disrupted, i.e., the crystalline domains should be converted into amorphous ones. For this purpose, several pretreatments of lignocellulosic materials have been extensively explored (e.g., chemical degradations, mechanical comminution, activation by swelling, ammonia fiber expansion and water-vapor explosion of the wooden fibers) [8,9]. As will be discussed later on, the dissolution of cellulose offers an alternative to the conventional pretreatments.
13.3 Cellulose in Solution
For nearly a century, processes to dissolve cellulose as well as regenerate the processed polymer from its solutions were developed to reshape the native polymer into fibers and films [10]. Nowadays, most of the manmade cellulose-based fibers are produced by the viscose and Lyocell processes. In the viscose process, cellulose becomes a soluble derivative in 5–8% aqueous NaOH solutions upon treating the fibers with
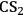 and NaOH [10]. In the Lyocell process, N-methylmorpholine-N-oxide (NMNO) is a non-derivatizing solvent that dissolves cellulose [10]. Despite efficacy, these solvents are expensive and not environmentally friendly. Thus, advances toward improved processes for the dissolution of cellulose are necessary not only for the production of fibers, but also in the biorefining of lignocellulose. In fact, solvents could play an important role in driving down the costs involved in the pretreatment of lignocellulose. Nowadays, the lignocellulose pretreatment (via two stage, dilute sulfuric acid hydrolysis [11]) accounts for about 20% of the direct costs involved in the production of cellulosic ethanol [9]. Considering enzymatic hydrolysis, the pretreatment costs can reach 40–50% of the final price of cellulosic ethanol [9]. In this scenario, improving the knowledge either in the dissolution or in the swelling of lignocellulose could provide the key to alternative, less expensive pretreatment processes. In this section, a discussion on the main aspects of the dissolution of cellulose is given.
and NaOH [10]. In the Lyocell process, N-methylmorpholine-N-oxide (NMNO) is a non-derivatizing solvent that dissolves cellulose [10]. Despite efficacy, these solvents are expensive and not environmentally friendly. Thus, advances toward improved processes for the dissolution of cellulose are necessary not only for the production of fibers, but also in the biorefining of lignocellulose. In fact, solvents could play an important role in driving down the costs involved in the pretreatment of lignocellulose. Nowadays, the lignocellulose pretreatment (via two stage, dilute sulfuric acid hydrolysis [11]) accounts for about 20% of the direct costs involved in the production of cellulosic ethanol [9]. Considering enzymatic hydrolysis, the pretreatment costs can reach 40–50% of the final price of cellulosic ethanol [9]. In this scenario, improving the knowledge either in the dissolution or in the swelling of lignocellulose could provide the key to alternative, less expensive pretreatment processes. In this section, a discussion on the main aspects of the dissolution of cellulose is given.
13.3.1 Physical Chemical Aspects of the Dissolution Process
The solubility of 1,4-β-glucans is related to their DP [5]. Oligomers composed of 2 to 6 AGUs are quite soluble in water, whereas molecules comprising 7 to 13 AGUs are partially soluble in hot water [5]. As the size of the cellulosic chain grows, packing up the polymeric chains by a dense network of intermolecular H-bonds becomes energetically favorable [12]. In general, microfibrils comprising polymeric chains greater than 30 AGUs are insoluble in most of the typical polar solvents and highly resistant to chemical and biological transformations, displaying the typical properties of cellulose [1,5].
The dissolution of cellulose begins with the molecules of solvent diffusing through the polymeric matrix. Accordingly, the polymer swells becoming highly solvated, forming a gel. However, the dissolution process is only complete when the structure of the gel is broken, dispersing the macromolecules into the solvent. It is important to keep in mind that a polymeric solution is indeed a colloidal system because of the dimensions of the macromolecule (e.g., a straight chain of cellulose comprising 200 AGUs measures ca. 100 nm in length [5]).
The dissolution of cellulose is a spontaneous process, at a given temperature, only if the free energy of solution,
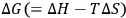 , is negative. The entropy of solution,
, is negative. The entropy of solution,
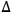 S, always assumes a positive value because of the increased conformational mobility of the polymeric chains in solution. Thus, the term “
S, always assumes a positive value because of the increased conformational mobility of the polymeric chains in solution. Thus, the term “
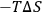 ” contributes toward an exergonic process (
” contributes toward an exergonic process (
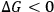 ). In this manner, the sign of
). In this manner, the sign of
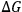 is determined by the enthalpy of solution, which is approximately equal to the heat of mixing. In turn, the heat of mixing,
is determined by the enthalpy of solution, which is approximately equal to the heat of mixing. In turn, the heat of mixing,
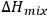 , is given by Equation 13.1:
, is given by Equation 13.1:
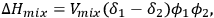
|
13.1 |
where
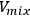 is the volume of the mixture,
is the volume of the mixture,
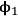 and
and
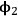 are the volume fractions of the two components, and δ is the solubility parameter (or also called Hildebrand parameter).
are the volume fractions of the two components, and δ is the solubility parameter (or also called Hildebrand parameter).
Analyzing Equation 13.1 shows that the term (
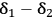 ) defines the extent to which
) defines the extent to which
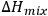 is a positive value. Hence, to obtain an enthalpy of mixing near to zero, it is required that
is a positive value. Hence, to obtain an enthalpy of mixing near to zero, it is required that
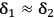 . Under this condition, solely entropic effects will govern the dissolution process. As a result, the free energy of solution will invariably assume a negative value, i.e., the dissolution is spontaneous at a given temperature.
. Under this condition, solely entropic effects will govern the dissolution process. As a result, the free energy of solution will invariably assume a negative value, i.e., the dissolution is spontaneous at a given temperature.
The δ parameter is defined by Equation 13.2:
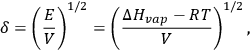
|
13.2 |
where
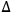 E and V stand for the energy of vaporization and the molar volume of the component respectively. Because
E and V stand for the energy of vaporization and the molar volume of the component respectively. Because
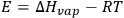 (where
(where
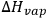 is the latent heat of vaporization, R is the gas constant, and T is the absolute temperature in Kelvin), δ can be directly determined from
is the latent heat of vaporization, R is the gas constant, and T is the absolute temperature in Kelvin), δ can be directly determined from
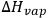 for volatile solvents.
for volatile solvents.
The term (E/V) is known as cohesive energy density. The physical meaning of this term is the energy required to remove a molecule from its nearest neighbor. For compounds having negligible vapor pressure such as cellulose, the cohesive energy density can be determined via direct and indirect methods (e.g., gas-solid chromatography [13], mechanical measurements [14], from calculations using group molar attraction constants [14] and from known relationships with the free energy of surface [14]).
To date, there has been little agreement on the values of the δ parameter of microcrystalline cellulose. The value determined by a direct method, based on inverse gas chromatography, is 39.9 MPa1/2 (Table 13.1, entry 34) [13]. Questions were raised about the method because the material needs to be preconditioned at 80 °C for 48 hours removing water from the surface of the cellulose [14]. An indirect mechanical measurement revealed a much lower value of the δ parameter, 25.7 MPa1/2 (Table 13.1, entry 35) [14]. Furthermore, the calculation of the δ parameter using the group molar attraction constants results in a value of 30.2 MPa1/2 (Table 13.1, entry 36) [14]. In turn, Hansen considered that amorphous cellulose would have a δ parameter close to the value found for Dextran C, 38.6 MPa1/2 (Table 13.1, entry 37) [15]. Finally, the solubility of cellulose in the ionic liquid 1-butyl-3-methylimidazolium chloride, [BMIM]Cl, suggests that the value of δ should be about 35.0 MPa1/2.
| Entry | Molecular solvents | δ | δP | δD | δH (MPa)1/2 | Ra(34) | Ra(36) | Ra(37) |
|---|---|---|---|---|---|---|---|---|
| 1 | nHexane | 14.9 | 14.9 | 0 | 0 | 35 | 25.8 | 29.7 |
| 2 | Diethylether | 15.8 | 14.5 | 2.9 | 5.1 | 29.7 | 20.2 | 26.8 |
| 3 | Ethyl acetate | 18.1 | 15.8 | 5.3 | 7.2 | 26.2 | 17.7 | 23.2 |
| 4 | Toluene | 18.2 | 18.0 | 1.4 | 2.0 | 31.5 | 23.8 | 24.1 |
| 5 | Methyl ethyl ketone | 19.0 | 16.0 | 9.0 | 5.1 | 27.3 | 19.8 | 24.4 |
| 6 | Tetrahydrofuran | 19.4 | 16.8 | 5.7 | 8 | 24.9 | 17.0 | 21.1 |
| 7 | Cyclohexanone | 19.6 | 17.8 | 6.3 | 5.1 | 27.2 | 20.1 | 21.8 |
| 8 | Acetone | 20.0 | 15.5 | 10.4 | 7.0 | 25.6 | 18.2 | 23.9 |
| 9 | 1,4-Dioxane | 20.5 | 19.0 | 1.8 | 7.4 | 26.3 | 19.2 | 18.5 |
| 10 | Carbon disulfide | 20.5 | 20.5 | 0 | 0.6 | 33.3 | 26.8 | 23.2 |
| 11 | Acetic acid | 21.4 | 14.5 | 8.0 | 13.5 | 20.9 | 11.7 | 22.2 |
| 12 | Pyridine | 21.8 | 19.0 | 8.8 | 5.9 | 25.7 | 20.1 | 19.7 |
| 13 | N-Methyl-2-pyrrolidone | 22.9 | 18.0 | 12.3 | 7.2 | 24.3 | 19.0 | 19.9 |
| 14 | 2-Propanol | 23.5 | 15.8 | 6.1 | 16.4 | 17.8 | 8.4 | 18.5 |
| 15 | DMF | 24.8 | 17.4 | 13.7 | 11.3 | 20.4 | 15.5 | 17.9 |
| 16 | Formic acid | 24.9 | 14.3 | 11.9 | 16.6 | 17.9 | 10.1 | 21.6 |
| 17 | Ethanol | 26.5 | 15.8 | 8.8 | 19.4 | 14.5 | 5.8 | 17.8 |
| 18 | Dimethylsulfoxide | 26.7 | 18.4 | 16.4 | 10.2 | 21.5 | 18.2 | 17.1 |
| Entry | Molecular solvents | δ | δP | δD | δH (MPa)1/2 | Ra(34) | Ra(36) | Ra(37) |
|---|---|---|---|---|---|---|---|---|
| 19 | NMNO | 26.9 | 19.0 | 16.1 | 10.2 | 21.4 | 18.5 | 16.3 |
| 20 | Triethyleneglycol | 27.5 | 16.0 | 12.5 | 18.6 | 14.4 | 8.4 | 17.5 |
| 21 | Methanol | 29.6 | 15.1 | 12.3 | 22.3 | 12.5 | 6.2 | 19.0 |
| 22 | Diethyleneglycol | 29.9 | 16.2 | 14.7 | 20.5 | 12.7 | 9.0 | 16.7 |
| 23 | Propyleneglycol | 30.2 | 16.8 | 9.4 | 23.3 | 10.1 | 3.6 | 15.3 |
| 24 | Ethanolamine | 31.5 | 17.2 | 15.6 | 21.3 | 11.3 | 9.9 | 14.5 |
| 25 | Dipropyleneglycol | 31.7 | 16.0 | 20.3 | 18.4 | 16.5 | 14.9 | 17.5 |
| 26 | Ethyleneglycol | 32.9 | 17.0 | 11.0 | 26.0 | 7.4 | 5.0 | 15.3 |
| 27 | Glycerol | 36.1 | 17.4 | 12.1 | 29.3 | 4.6 | 7.7 | 15.6 |
| 28 | Formamide | 36.6 | 17.2 | 26.2 | 19.0 | 18.8 | 20.4 | 14.9 |
| 29 | Water | 47.8 | 15.6 | 16.0 | 42.3 | 13.8 | 19.8 | 26.7 |
| Ionic liquids | ||||||||
| 30 | [BMIM]PF6 | 29.3 | 21.0 | 17.2 | 10.9 | 21.1 | 20.2 | 13.4 |
| 31 | [OMIM]PF6 | 27.8 | 20.0 | 16.5 | 10.0 | 21.7 | 19.6 | 15.2 |
| 32 | [BMIM]BF4 | 31.5 | 23.0 | 19.0 | 10.0 | 23.3 | 24.0 | 13.1 |
| 33 | [BMIM]Cl | 35.0 | 19.1 | 20.7 | 20.7 | 13.3 | 15.9 | 10.6 |
| Substrates | R0 | |||||||
| 34 | Cellulose | 39.3 | 19.4 | 12.7 | 31.3 | - | ||
| 35 | Cellulose | 24.7 | - | - | - | - | ||
| 36 | Cellulose | 30.2 | 15.8 | 6.8 | 24.8 | - | ||
| 37 | Dextran C | 38.6 | 24.3 | 19.9 | 22.5 | 17.4 |
Tab. 13.1: Solubility parameters of selected solvents and polysaccharides [13-18]
Tab. 13.1: Solubility parameters of selected solvents and polysaccharides [13-18]
Considering 39.9 MPa1/2 as the value of the δ parameter of cellulose, the analysis of Table 13.1 reveals that only the molecular solvent, formamide (Table 13.1, entry 26), and the ionic liquid [BMIM]Cl (Table 13.1, entry 26) would have similar solubility parameters to cellulose. Formamide does not dissolve cellulose, whereas [BMIM]Cl does. Actually, the Hildebrand parameter often fails in the prediction of the solubility of highly H-bonded compounds such as cellulose [17].
The cohesive energy,
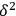 , is indeed the sum of three distinct cohesive energies resulting from (1) nonpolar, atomic (dispersion) interactions,
, is indeed the sum of three distinct cohesive energies resulting from (1) nonpolar, atomic (dispersion) interactions,
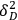 , (2) permanent dipole-dipole molecular interactions,
, (2) permanent dipole-dipole molecular interactions,
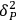 , and (3) hydrogen bonding interactions,
, and (3) hydrogen bonding interactions,
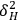 , as indicated in Equation
, as indicated in Equation
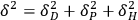
|
13.3 |
The solubility parameters δD, δP and δH are known as Hansen Solubility Parameters or HSP. Analyzing separately the terms that contribute to the overall cohesive energy, δ2, helps to understand why solvents having the same value of δ parameter, such as 1,4-dioxane and
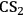 (Table 13.1, entries 9 and 10), sometimes possess totally different properties. In this case, the term δH clearly differentiates 1,4-dioxane from
(Table 13.1, entries 9 and 10), sometimes possess totally different properties. In this case, the term δH clearly differentiates 1,4-dioxane from
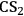 .
.
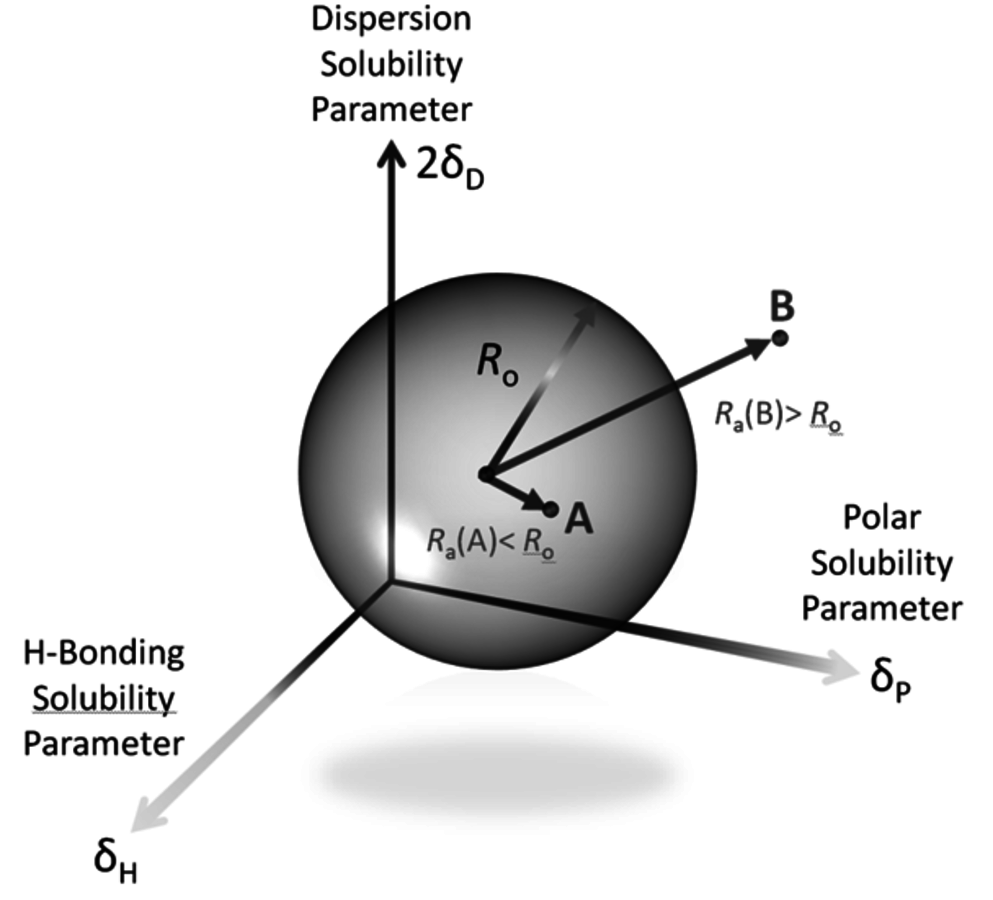
Fig. 13.3: Three-dimensional visualization of the Hansen Solubility Parameters (HSP) in the characterization of a good solvent (A) and a poor solvent (B). Adapted from [17]
According to Hansen [17], dissolution or swelling takes place when a solvent has the values of δD, δP and δH close to those of the polymer. Figure 13.3 shows a three-dimensional representation of the HSP characterization. The HSP of the polymer are at the center of the sphere. The radius of the sphere, Ro, indicates the maximum difference in affinity that is tolerable for a “good” interaction between solvent and solute. This parameter is empirically determined by comparing the solubility of a solute in several solvents [15]. Good solvents are within the sphere, whereas the poor ones are outside. For instance, solvent A is a good solvent for the solute, but solvent B does not have the adequate HSP, as illustrated in Figure 13.3 [15].
The dissolution or the swelling of the polymer is predicted to occur if the condition
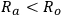 is satisfied (Figure 13.3). The interaction distance between the solvent and the center of the sphere, Ra, is calculated as indicated in Equation 13.4 [15,17]:
is satisfied (Figure 13.3). The interaction distance between the solvent and the center of the sphere, Ra, is calculated as indicated in Equation 13.4 [15,17]:
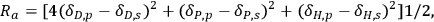
|
13.4 |
where the letters p and s are the descriptors for the HSP values of the polymer and the solvent, respectively.
Table 13.1 shows the interaction distances Ra (entry 34), Ra (entry 36) and Ra (entry 37) that were calculated considering the HSP values of microcrystalline cellulose (entries 34 and 36) and dextran C (entry 37). It is clear from Table 13.1 that [BMIM]Cl is one of the solvents with a short interaction distance. However, the solvents listed in the entries 21 to 24, 26 and 27 have shorter distance values than the one found for [BMIM]Cl. Although cellulose swells in some of these solvents, they are not capable of dissolving it. Considering that the HSP values of amorphous cellulose are the same as those of dextran C [15], a better differentiation of solvents can be achieved using a value of Ro equal to 17.4. Due to the structural differences between cellulose and dextran C, caution must be applied in this analysis as the HSP values of dextran C might not be transferable to amorphous cellulose. For example, the ionic liquids assume quite similar values of Ra, but only [BMIM]Cl dissolves cellulose.
The HSP is a thermodynamic approach to predict solubility. It may happen that the prediction indicates that the material is soluble in a solvent, but in reality the material is only swollen or even insoluble in the solvent. Several reasons may account for the “incorrect” prediction. First, if the solvent molecules are too large to penetrate the pore structure of the polymer, the swelling of the polymer as well as its dissolution will be extremely slow. Second, although
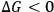 implies that the process is spontaneous, this gives no information about the rate of the process, i.e., a spontaneous process may never happen due to a kinetic barrier between the initial and the final states. Third, the HSP approach considers a solution as an ideal mixture. Thus, specific interactions between a solvent and a polymer are not considered in the analysis. Finally, the prediction may be false. This may be the case for cellulose because of the supposed inaccuracy of the HSP determined for this polymer.
implies that the process is spontaneous, this gives no information about the rate of the process, i.e., a spontaneous process may never happen due to a kinetic barrier between the initial and the final states. Third, the HSP approach considers a solution as an ideal mixture. Thus, specific interactions between a solvent and a polymer are not considered in the analysis. Finally, the prediction may be false. This may be the case for cellulose because of the supposed inaccuracy of the HSP determined for this polymer.
| Class | Type | Examples |
|---|---|---|
| Mineral acids | Concentrated mineral acids | H2SO4, HCl, HF, H3PO4 |
| Aqueous systems | Transition metal complexes containing NH3 and/or amine ligands (excess of NH3 or amine is required) | Cadoxen – [Cd(H2N(CH)2NH2)3](OH)2, Cupren – [Cu(H2N(CH)2NH2)2](OH)2, Cuam – [Cu(NH3)4](OH)2, Zincoxen – [Zn(H2N(CH)2NH2)2](OH)2 |
| Transition metal tartrates | FeTNa – NA6[Fe(C4H3O6)3] | |
| Quaternary ammonium hydroxides | Triton B, TEOH, Triton F, Guanidinium hydroxide | |
| Alkali hydroxides | NaOH, LiOH | |
| Tertiary amines oxides | N-Methylmorpholine-N-oxide | |
| Molten salt hydrates | Swelling agents | LiCl•xH2O (2 ≤ x ≤ 5), Zn(NO3)2•6H2O, NaClO4•H2O, Mg(ClO4)2•H2O, LiClO4•3H2O/CaCl2±6H2O |
| Solvent media | ZnCl2•4H2O, LiClO4•3H2O, Zn(NO3)2•xH2O (x < 6), FeCl3•6H2O, LiSCN•2H2O, LiI•2H2O, LiClO4•3H2O/MgCl2•6H2O, LiClO4•3H2O/Mg(ClO4)2/H2O, LiClO4•3H2O/NaClO4/H2O, LiCl/ZnCl2/H2O, NaSCN/KSCN/LiSCN/H2O | |
| Non-aqueous systems | Alkyl imidazolium ionic liquids |
[EMIM]Cl: 1-ethyl-3-methylimidazolium chloride, [BMIM]Cl: 1-butyl-3-methylimidazolium chloride |
| Class | Type | Examples |
|---|---|---|
| Non-aqueous systems | Alkyl imidazolium ionic liquids |
[EMIM][AcO]: 1-ethyl-3-methylimidiazolium acetate, [EMIM][OP(O)(OMe)2]: 1-ethyl-3-methylimidazolium dimethylphosphate |
| Non-aqueous systems | N Alkylpyridinium salts | N-Ethylpyridinium chloride |
| Tertiary amine oxides | N-Methylmorpholine-N-oxide, Triethylamine-N-oxide, N-Methylpiperidine-N-oxide | |
| DMSO based-solvent media |
DMSO/methylamine, DMSO/KSCN, DMSO/CaCl2, DMSO/tetrabutylammonium fluoride (TBAF) | |
| Liquid NH3 based-solvent media | NH3/NaI(NH4I), NH3/NaSCN(NH4SCN) | |
| Dipolar aprotic solvents/LiCl | N,N-Dimethylacetamide/LiCL, N-Methylpyrrolidone/LiCl | |
| Tricomponent solvent media | NH4/NaCl/DMSO, Ethylenediamine/NaI/N,N-Dimethylformamide, DIethylamine/SO2/DMSO |
Tab. 13.2: Some solvents for cellulose. Adapted from [10,19-21]
Tab. 13.2: Some solvents for cellulose. Adapted from [10,19-21]
13.3.2 Solvents for Cellulose
There are several solvents or solvent systems that fulfill the thermodynamic and kinetic requirements to dissolve cellulose. A short list of solvent systems for cellulose is given in Table 13.2 [10,19-21].
1Cellulose acts as a base (when solvents such as
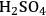 , HCl or
, HCl or
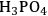 are employed);
are employed);
2Cellulose acts as an acid (when solvents such as urea/NaOH/water or ionic liquids are used);
3Cellulose acts as a chelating agent (when solvents such as cadoxen or cupram are applied);
4Cellulose is converted into a soluble derivative (such as in the viscose process in which the soluble polymer is the water-soluble cellulose xanthanate).
Figure 13.4 illustrates the roles of cellulose in solution. The terms base and acid describe cellulose as an H-bond acceptor or as an H-bond donor, respectively. Considering cellulose as a chelating agent, the ΔGreaction related to the complex formation also gives an important contribution to the overall ΔGsolution.
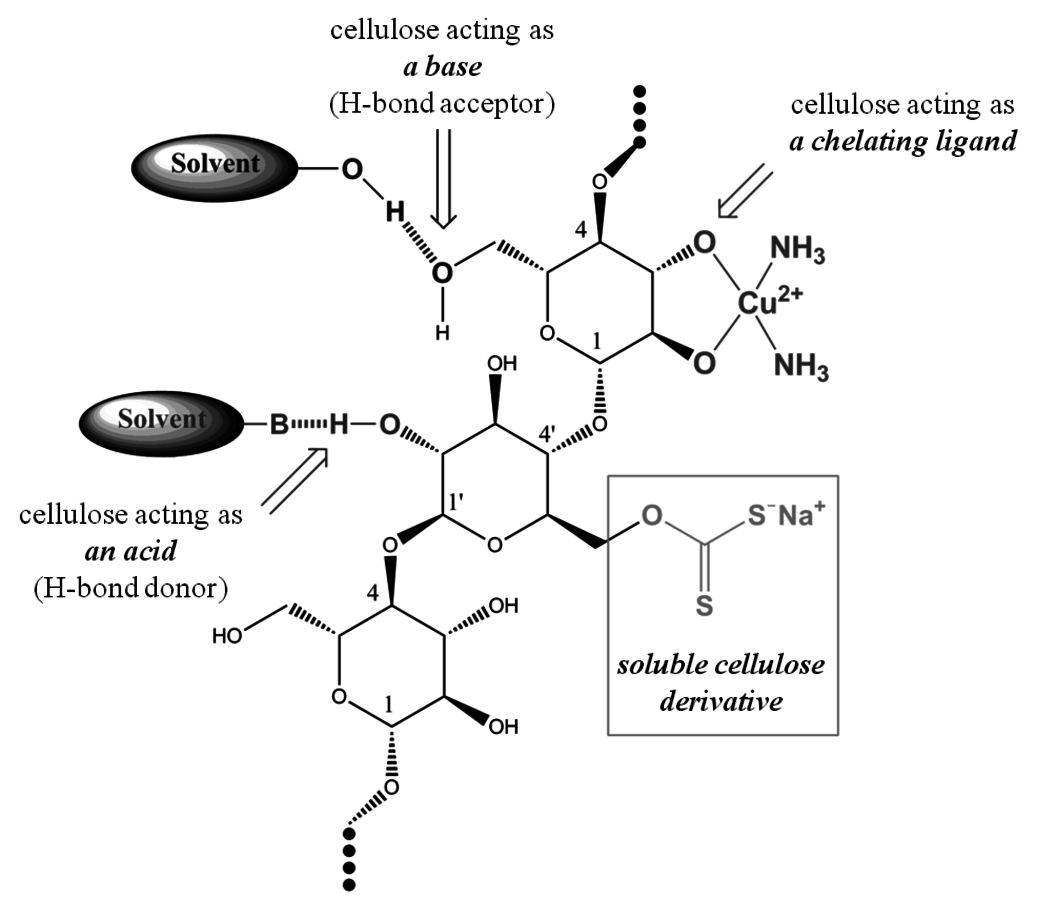
Fig. 13.4: The roles of cellulose in solution [21]
Although the prospect of producing manmade fibers from cellulose previously dominated the research and development in solvents [10], recent efforts have focused on the development of solvent systems for the processing of lignocellulose. The use of ionic liquids (ILs) as solvents for cellulose is the most important example in the current RD [18,22,23]. ILs are salts that melt below 100 °C [22]. The 1-alkyl-3-methylimidazolium-based ILs comprising good H-bond acceptor anions (e.g.,
 ,
,
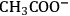 and
and
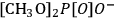 to mention some [24,25]) can dissolve cellulose [22,26] and even wood [18,23]. However, the high cost related to the use of ILs still hinders their applicability in biorefineries. Moreover, the recycling of ILs is a big challenge due to the lack of efficient separation processes to extract highly polar compounds (e.g., sugars or 5-hydroxymethylfurfural) from ILs [18,23].
to mention some [24,25]) can dissolve cellulose [22,26] and even wood [18,23]. However, the high cost related to the use of ILs still hinders their applicability in biorefineries. Moreover, the recycling of ILs is a big challenge due to the lack of efficient separation processes to extract highly polar compounds (e.g., sugars or 5-hydroxymethylfurfural) from ILs [18,23].
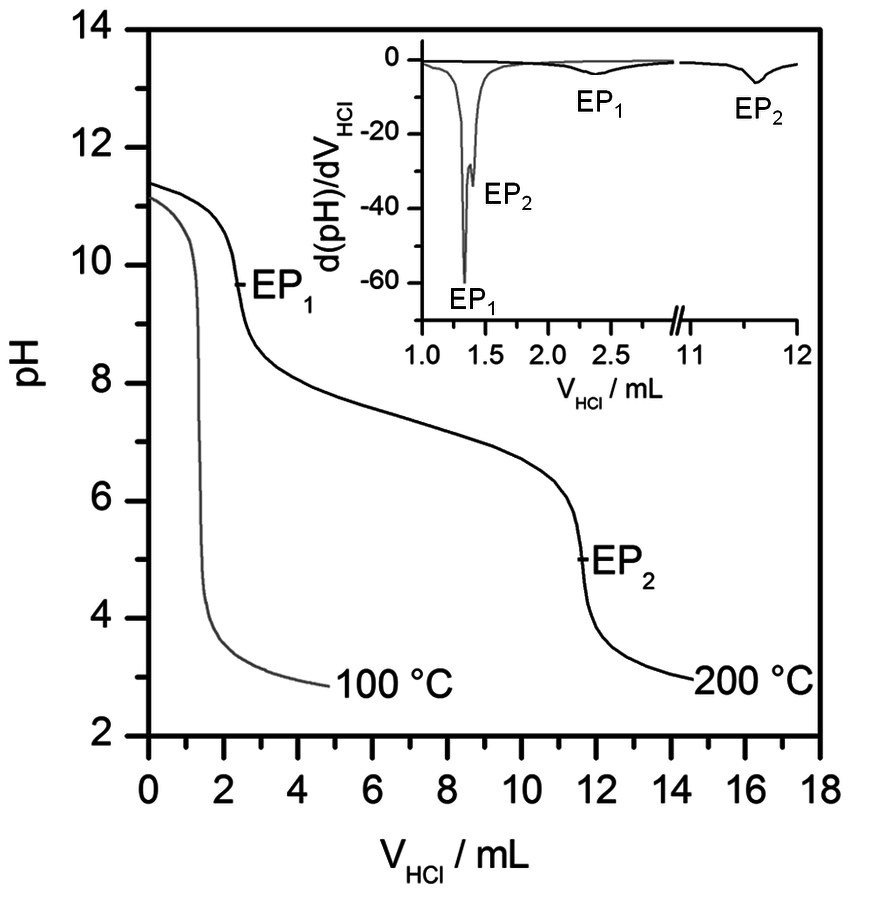
Fig. 13.5: Back-titration curves obtained from the samples of [BMIM]Cl heated at 100 and 200 °C for 24 hours [27]
In process development, primary criteria for the selection of ILs are not only price, toxicity and recyclability, but also the degradation temperature of the IL is important in the final decision. The first serious discussions on the thermal stability of ILs emerged from investigations using thermogravimetric analysis (TGA) [28]. Although efforts to determine the degradation temperature of several ILs were undertaken, the reported data are rather controversial, and there is no general agreement about the published degradation temperatures for the same ionic liquid [28]. For instance, degradation temperatures between 235 and 450 °C are published for 1-methyl-3-butylimidazolium bis(trifluoromethylsulfonyl)imide [28].
Recently, the potentiometric titration of alkylimidazoles was used to assess the deterioration of the ILs’ quality occurring upon thermal aging [27]. Figure 13.5 shows the curves of back-titration obtained from the samples of [BMIM]Cl heated at 100 and 200 °C for 24 hours. The first equivalent point, EP1, is due to the neutralization of the excess of NaOH solution dosed in the direct titration. The second equivalent point,
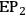 , is associated with the amount of free imidazoles in the samples.
, is associated with the amount of free imidazoles in the samples.
The potentiometric titration of the [BMIM]Cl sample treated at 200 °C for 24 hours revealed decomposition of ILs already at much lower temperatures than those inferred from TGA [27]. Table 13.3 lists the imidazole titers found in the untreated samples of some ILs and in those heated at 200 °C for 24 hours [27].
| IL | Tonset (°C) | Imidazoles (μmol g-1) as purchased |
Imidazoles (μmol g-1) 200 °C |
|---|---|---|---|
| [EMIM]Cl | 222 | 16 ± 1 | 332 ± 4 |
| [BMIM]Cl | 214 | 7 ± 1 | 874 ± 10 |
| [BMIM][CH3SO3] | 295 | 14 ± 1 | 31 ± 1 |
| [BMIM][Tf2N] | 367 | 5 ± 1 | 21 ± 2 |
Tab. 13.3: Thermal stability of IL samples aged at 200° C for 24 hours [27]. The onset decomposition temperature, Tonset, was determined by TGA [27].
The long-term stability of ILs is an important issue for industrial use. Figure 13.6 shows the evolution of the imidazole content in [BMIM]Cl (99.9%) samples heated at 100 or 140 °C for 10 days. Although the [BMIM]Cl samples heated at 100 °C show no degradation over 10 days, the imidazoles content increases from 13 to 62 µmol g-1 after heating the samples over 10 days at 140 °C under anhydrous conditions. On the other hand, a titer of 78 µmol g-1 was found upon aging a sample containing 2 wt% water at 140 °C for 10 days. This shows that water also affects the ionic liquid thermal stability [27].
The solubility of cellulose in ILs is another important aspect to be considered. Currently, the ILs available in the market cannot dissolve more than 25 wt% of cellulose, which results in very viscous solutions or even “sticky” gels [18,22,26]. Furthermore, a typical difficulty experienced while dissolving cellulose in ILs is the agglomeration of the cellulosic fibers. The resulting clumps take very long to dissolve. Slowly pouring the biopolymer into the IL under vigorous mechanical stirring helps to break up the clumps of cellulose to a certain extent, however, this unit operation is rather energy demanding.
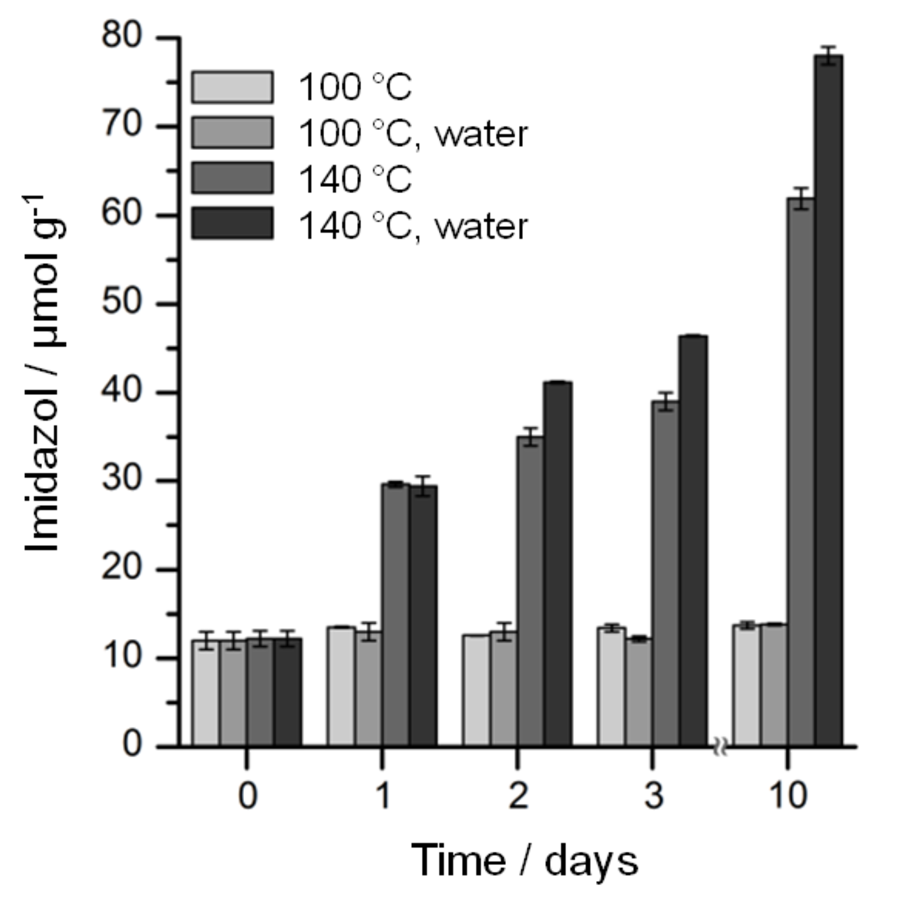
Fig. 13.6: Effect of water (2 wt%) on long-term stability of [BMIM]Cl samples heated at 100 and 140 °C [27]
Recently, solvent systems comprising IL and molecular solvents were reported [29]. They circumvented some of the most difficult hurdles faced when using neat ILs. For instance, cellulose clumps are not formed, the dissolution takes place quickly, and the amount of ILs required for the process can be markedly reduced [29].
Figure 13.7 shows the effect of the concentration of [BMIM]Cl on the dissolution of microcrystalline cellulose (Avicel, 10 wt%) dispersed in 1,3-dimethyl-2-imidazolidinone (DMI). The dissolution process starts upon the addition of [BMIM]Cl into the suspension. A white paste is obtained in the system containing 10 wt% IL. The increase of IL concentration from 20 to 40 wt% makes the mixture gradually more transparent. Cellulose dissolves completely in a 50 wt% solution of [BMIM]Cl in DMI after only 3 minutes [29]. For the sake of comparison, the regular dissolution of cellulose in neat [BMIM]Cl typically takes more than 10 hours to complete [22].

Fig. 13.7: Appearance of the mixtures comprising microcrystalline cellulose (Avicell), DMI and [BMIM]Cl (0-50 wt%) after stirring at 100 °C for the time indicated in the figure [29]
Instantaneous dissolution of cellulose at 100 °C is achieved when [BMIM]Cl is replaced by 1-ethyl-3-methylimidazolium acetate, [EMIM][AcO]. Figure 13.8 shows some examples of IL-based electrolyte solutions capable of dissolving cellulose instantaneously at 100 °C [29].
The mole fraction of [EMIM][AcO], χ[EMIM][AcO], required for the dissolution of cellulose depends on the molecular solvent used in the electrolyte solution. For several of the reported amide-containing solvents, the dissolution takes place even when χ[EMIM][AcO] is lower than 0.30. The DMSO/[EMIM][AcO] system requires the smallest χ[EMIM][AcO] (0.09). On the other hand, the dissolution of cellulose in acetylacetone, tert-butanol and tert-pentanol happens only when an equimolar amount of IL is present in the mixture. Surprisingly, N,N,N,N-tetramethylurea, an analogue of DMI, requires the largest χ[EMIM][AcO] (0.59) for the dissolution of cellulose [29].
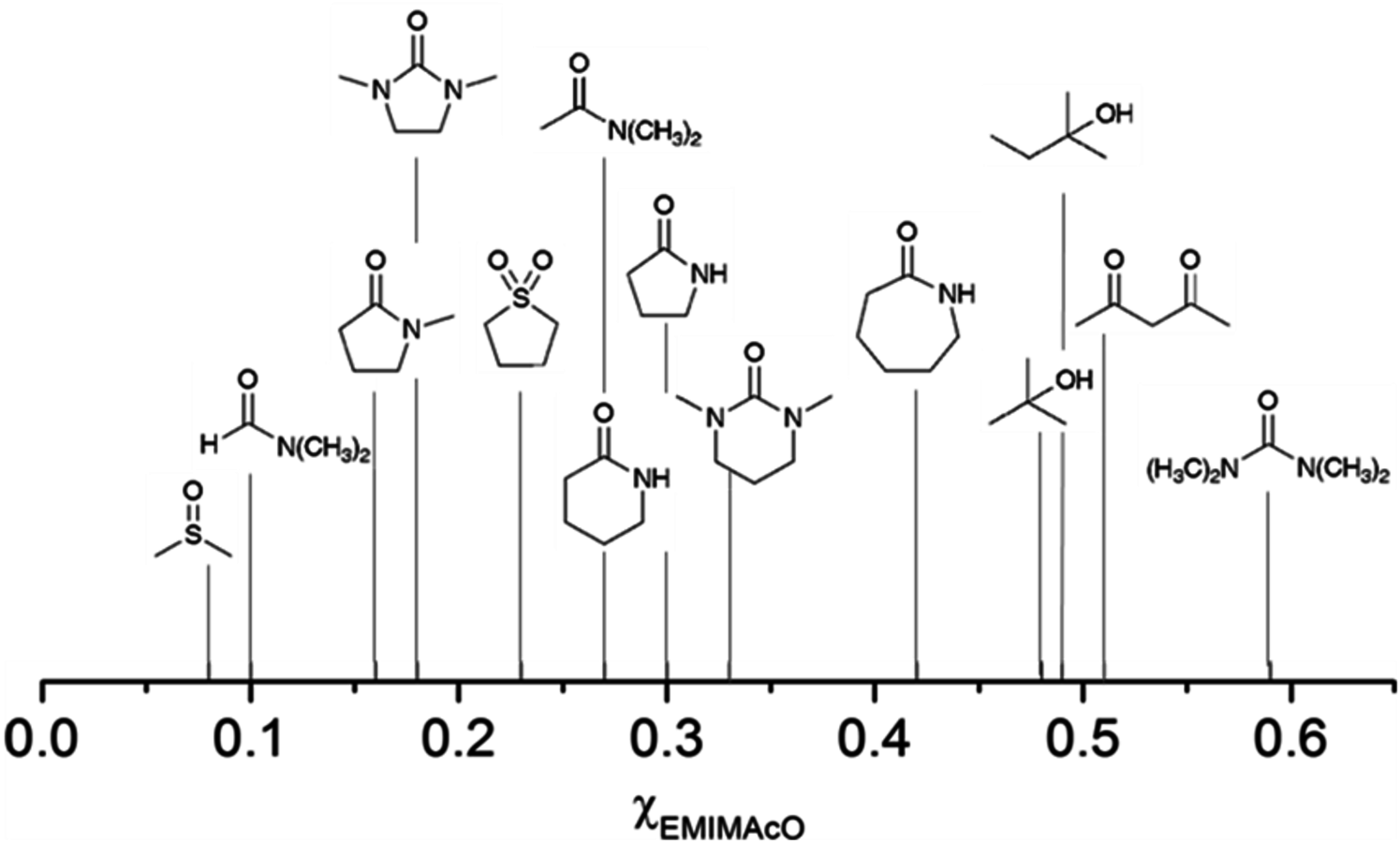
Fig. 13.8: The mole fraction of [EMIM][AcO] required for the dissolution of microcrystalline cellulose (10 wt%) in several molecular solvents at 100 °C. Adapted from [29].
The concept of distillable acid-base conjugate ILs was recently introduced [30]. Cellulose (10 wt%) is soluble in

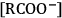 , where
, where
 stands for 1,1,3,3-tetramethylguadinidium and R is -H, -
stands for 1,1,3,3-tetramethylguadinidium and R is -H, -
 and -
and -
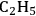 . These guanidinium-based ionic liquids can be distillated in a Kügelrohr short-path distillation apparatus (3 g, 100 to 200 °C over 30 minutes, 1 mmHg) [27]. This is possible because of the equilibrium:
. These guanidinium-based ionic liquids can be distillated in a Kügelrohr short-path distillation apparatus (3 g, 100 to 200 °C over 30 minutes, 1 mmHg) [27]. This is possible because of the equilibrium:

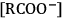
 TMG + RCOOH, where TMG stands for 1,1,3,3-tetramethylguadinidine. Above 100 °C and under vacuum, the equilibrium shifts towards the molecular compounds enabling the distillation. At room temperature, the molecular compounds recombine regenerating the IL.
TMG + RCOOH, where TMG stands for 1,1,3,3-tetramethylguadinidine. Above 100 °C and under vacuum, the equilibrium shifts towards the molecular compounds enabling the distillation. At room temperature, the molecular compounds recombine regenerating the IL.
13.4 Homogeneous Hydrolysis of Cellulose
Hydrolysis of cellulose is typically performed under heterogeneous reaction conditions, i.e., the substrate is not dissolved in the reaction medium [5]. However, the solvent does not function merely as a dispersant. Cellulose can also be swollen in the solvent. This activates the substrate. In heterogeneous reactions, the chemical transformation of cellulose occurs first on its surface [5]. Soluble intermediates, formed by the attack of the surface, undergo further reactions in solution, yet the complete conversion of cellulose is often not achieved in heterogeneous reactions. Hence, very recalcitrant cellulose accumulates in the process raising difficulties for catalyst recovery [2,5]. In the homogeneous hydrolysis of cellulose, the substrate is soluble in the reaction medium. Under this condition, the physical barriers to efficient hydrolysis are no longer present (e.g., crystallinity, morphology, surface area and other physical features) [5]. In this manner, the hydrolysis rate is several orders of magnitude higher than that found for the heterogeneous hydrolysis. Moreover, the integral utilization of cellulose can be achieved with ease [5]. As described in the previous section, several solvents for cellulose are available, but the choices become scarce when one envisages carrying out catalytic reactions in these media. In this section, the experience gained in the hydrolysis of cellulose in solutions of concentrated mineral acids [11,31-34], melt salt hydrates [35,36], and ionic liquids [5,23] is briefly described.
Hydrolysis of cellulose in concentrated mineral acids was one of the first methods used in the investigation of the composition of plant tissues. Anselme Payen in the 1830s discovered that the extracts, obtained from acid hydrolysis of oak and beech wood, have an elemental composition similar to that of starch [5]. Currently, the composition of lignocellulosic material is determined by the quantitative saccharification in sulfuric acid [37]. In the first stage, this method employs the dissolution and hydrolysis of cellulose (and hemicelluloses) in 72 wt% sulfuric acid at room temperature. In the second stage, the complete saccharification is achieved in dilute acid solution (3 wt%) at 130 °C. Monosaccharides are obtained as main products. Despite efficacy, this method is not feasible on large scale due to problems with corrosion and acid recycling.
The saccharification of wood in fuming hydrochloric acid (40 wt%), known as Bergius process, is the only process employing concentrated mineral acid that so far has been applied on a large scale [31]. Cellulose is soluble in fuming hydrochloric acid at room temperature. Under these conditions, the biopolymer hydrolyses to oligosaccharides within a few hours without considerable formation of dehydration products (e.g., furfural and 5-hydroxymethylfurfural [5-HMF]) [31]. In practice, after the removal of hydrochloric acid, about 1 wt% of the acidic content still remains in the raw hydrolyzate. The residual acidic content catalyzes the hydrolysis of the oligosaccharides to fermentable sugars, which is performed in a second stage at 120 °C for 0.5 hours. By the Bergius process, a ton of dry wood was claimed to yield 320 L of 95% ethanol [31]. Fuming hydrochloric acid shows two major advantages over sulfuric acid. First, hydrochloric acid permeates better than sulfuric acid into the wood fibers. Second, up to 99% of the hydrochloric acid can be recycled. However, the main economical drawbacks of this technology are the requirement of corrosion-resistant plants, and the expensive recovery of hydrochloric acid [38].
Concentrated phosphoric (85%) acid dissolves considerable amounts of cellulose (e.g., 20 wt% cotton linter) [34]. Moreover, cellulose controllably depolymerizes in this solvent. This method was first explored to produce low crystallinity, low DP celluloses for pharmaceutical purposes. The decay rate of DP is enhanced from 4.79×10-3h-1 at 25 °C to 0.314 h-1 at 50 °C. The activation energy of the reaction corresponds to a value of 106 kJ mol-1; this value is, for example, 20 to 40 kJ mol-1 lower than those found for the heterogeneous hydrolysis of cellulose [5,34]. Recently, mixtures comprising 70% phosphoric acid and 30% sulfuric acid were employed as reaction media for hydrolysis of cellulose [32]. The load of substrate (corn cob) was 30 wt%. This was slowly mixed in a blender at 30 °C. The initial thick mixture was left to decrystallize for 16 hours. Finally, water was added and the mixture was heated at 80 °C. The process led to about 90% sugar yield after 4 hours [32].
Several molten salt hydrates (Table 13.2) are good solvents for cellulose. Homogeneous hydrolysis of cellulose is reported to take place at 85 °C in zinc chloride tetrahydrate (using 0.4 molal of hydrochloric acid as catalyst) [35,36]. The reaction achieves a 70% yield of glucose after 0.5 hours [36]. Interestingly, salts cannot only dissolve cellulose, but they can also swell it [33]. In the heterogeneous hydrolysis of cellulose, the presence of
 (1.6 mol L-1) or LiCl (6 mol L-1) was shown to be beneficial in the hydrolysis of cellulose in hydrochloric acid (6–7 mol L-1, 7 wt%) at 90 °C. Yields up to 85% glucose are claimed [33]. The swelling effect of the salts in the cellulosic fibers is pointed out as a factor accounting for the enhancement of the hydrolysis rate [33].
(1.6 mol L-1) or LiCl (6 mol L-1) was shown to be beneficial in the hydrolysis of cellulose in hydrochloric acid (6–7 mol L-1, 7 wt%) at 90 °C. Yields up to 85% glucose are claimed [33]. The swelling effect of the salts in the cellulosic fibers is pointed out as a factor accounting for the enhancement of the hydrolysis rate [33].
Ionic liquids are the most explored solvents for the hydrolysis of cellulose. Recent reviews outline in detail the progress in the field [5,18,23,39]. The first report on hydrolysis of cellulose dissolved in [BMIM]Cl was given by Li and Zhao in 2007 [40]. They demonstrated that cellulose dissolved in [BMIM]Cl hydrolyzes in the presence of catalytic amounts of sulfuric acid or other mineral acids at 100 °C. A typical problem found in this reaction is the degradation of glucose into several products (Figure 13.9). Recently, a solution for this problem was given [41]. The selective production of glucose can be achieved upon controllably adding small amounts of water into the mixture over the course of the reaction [41]. Nonetheless, the separation of the highly polar products from the IL is still a problem without a practical solution.
The activation of cellulose toward hydrolysis requires a strong acid [5,42]. This prohibits the utilization of acetate- or phosphonate-based ionic liquids—or any other kind of ionic liquid comprising a weakly basic anion. These anions capture the
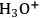 species preventing the activation of the glycosidic bonds. Additionally, the presence of N-methylimidazole, often found at different concentration levels as an impurity in [BMIM]Cl, decreases proportionally the catalytic performance of this system [42].
species preventing the activation of the glycosidic bonds. Additionally, the presence of N-methylimidazole, often found at different concentration levels as an impurity in [BMIM]Cl, decreases proportionally the catalytic performance of this system [42].
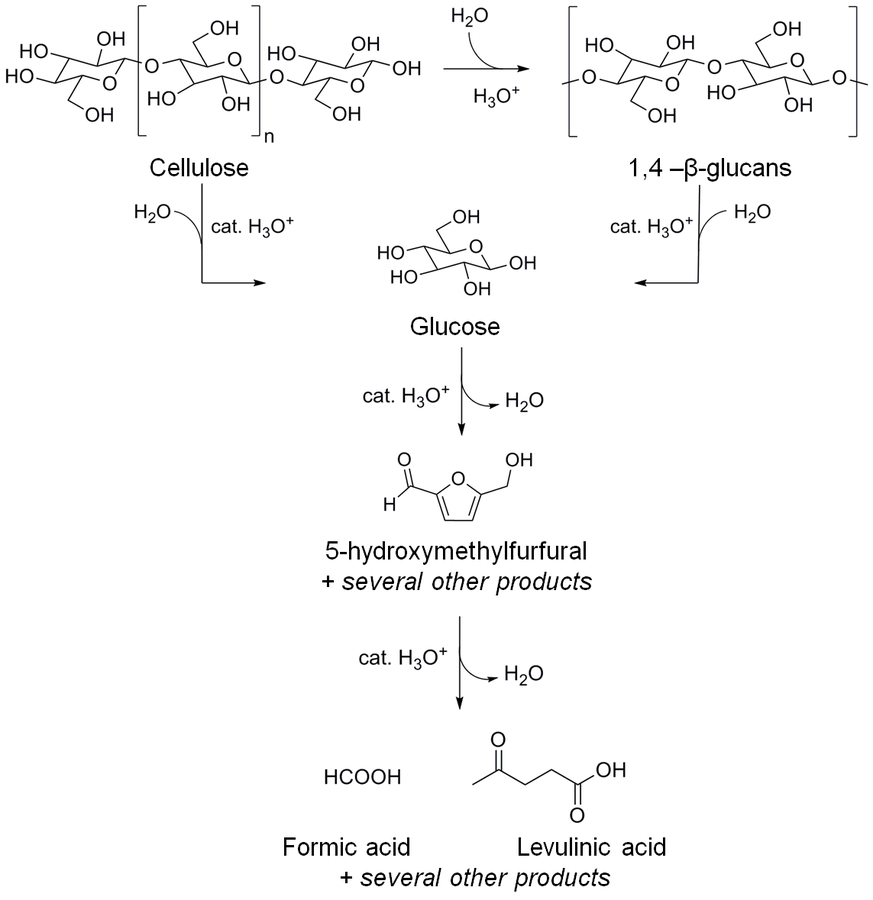
Fig. 13.9: Selected products formed by acid-catalyzed reactions starting from cellulose. As hemicelluloses are typical impurities of commercial celluloses, xylose and other monosaccharides as well as their degradation products (furfural, furoic acid, and others) may be also present in the reaction mixture [42].
The addition of
 species into a solution of cellulose in [BMIM]Cl initiates a complex reaction chain, as illustrated in Figure 13.9 [42]. In the first step, cellulose undergoes depolymerization via hydrolysis of 1,4-β-glycosidic bonds. Either smaller 1,4-β-glucans (cellooligomers) or glucose can be formed at this stage. Glucose is likely to be dehydrated under acidic conditions yielding several compounds (e.g., 5-HMF, levulinic acid, formic acid and several others) [42]. These products are prone to recombine with sugars or oligosaccharides via aldol-condensation, resulting in polymers with undefined structure and stoichiometry called humins [42].
species into a solution of cellulose in [BMIM]Cl initiates a complex reaction chain, as illustrated in Figure 13.9 [42]. In the first step, cellulose undergoes depolymerization via hydrolysis of 1,4-β-glycosidic bonds. Either smaller 1,4-β-glucans (cellooligomers) or glucose can be formed at this stage. Glucose is likely to be dehydrated under acidic conditions yielding several compounds (e.g., 5-HMF, levulinic acid, formic acid and several others) [42]. These products are prone to recombine with sugars or oligosaccharides via aldol-condensation, resulting in polymers with undefined structure and stoichiometry called humins [42].
Performing the acid-catalyzed depolymerization of cellulose using Amberlyst 15DRY, instead of molecular acids, enables the direct control of the reaction progress [42,43]. The initial rate of depolymerization is controlled by the slow release of
 species into the reaction medium. As the reaction proceeds at a slow rate, a preferential cleavage of large polymeric molecules is detected in the beginning of the reaction. Hence, cellooligomers with a tunable DP can be conveniently produced. Furthermore, the reaction can be also conducted to quantitatively produce water-soluble products. In practice, however, it is much more interesting to stop the process at the stage in which cellooligomers are the main products because the work-up for the extraction of sugar and dehydration products is commonly very difficult—or even unachievable—due to the high solubility of these products in ILs [5,43]. Cellooligomers can be separated as main products by adding water (Figure 13.10), methanol, dichloromethane or liquid ammonia into the reaction mixture [5].
species into the reaction medium. As the reaction proceeds at a slow rate, a preferential cleavage of large polymeric molecules is detected in the beginning of the reaction. Hence, cellooligomers with a tunable DP can be conveniently produced. Furthermore, the reaction can be also conducted to quantitatively produce water-soluble products. In practice, however, it is much more interesting to stop the process at the stage in which cellooligomers are the main products because the work-up for the extraction of sugar and dehydration products is commonly very difficult—or even unachievable—due to the high solubility of these products in ILs [5,43]. Cellooligomers can be separated as main products by adding water (Figure 13.10), methanol, dichloromethane or liquid ammonia into the reaction mixture [5].

Fig. 13.10: Hydrolysis of microcrystalline cellulose. Appearance of the cellulose recovered from [BMIM]Cl by addition of water. The values between parentheses represent the percentage of isolated cellulose [43].
Cellooligomers regenerated from IL comprises up to 95 wt% water. The high degree of swelling suggests that the cellulosic chains are highly accessible (Figure 13.11). Indeed, starting from the cellooligomers almost quantitative conversion of cellulose into fermentable sugars is achieved in the enzymatic catalysis in aqueous medium, as shown in Figure 13.12 [44].

Fig. 13.11: Making cellulose accessible. Both flasks contain the equivalent to 1 g of dry cellulose [44].
Figure 13.12 shows the performance of a commercial cellulase preparation (Celluclast®, T. reesei) in the enzymatic hydrolysis of several cellulosic materials. The untreated α-cellulose and cellulose regenerated from ILs show substrate conversions of 46 and 79%, respectively. In contrast, the cellooligomers obtained from the acid-catalyzed depolymerization are, in the best cases, nearly quantitatively hydrolyzed (94%) by cellulases within 4 hours.
Furthermore, this reaction produces exclusively cellobiose and glucose, maintaining the enzymatic selectivity [44]. Surprisingly, no marked effect of the DP on the enzymatic performance is observed in the reactions carried out with cellooligomers smaller than 800 anhydroglucose units (AGU). This finding has important implications for process development. Starting from native cellulose (DP between 2,000 and 10,000 AGU), suitable cellooligomers for the enzymatic hydrolysis would be already produced after 3 to 16 scissions of the cellulosic chains, which is reached in less than 1 hour through the acid-catalyzed depolymerization in IL. As cellooligomers are preferably produced in the earlier stages of acid-catalyzed depolymerization, losses of glucose, due to its dehydration, are completely suppressed. Consequently, the integration of acid- and enzyme-catalyzed conversion unites the advantages of both “worlds” bringing rapid and quantitative conversion of cellulose to fermentable sugars [44].
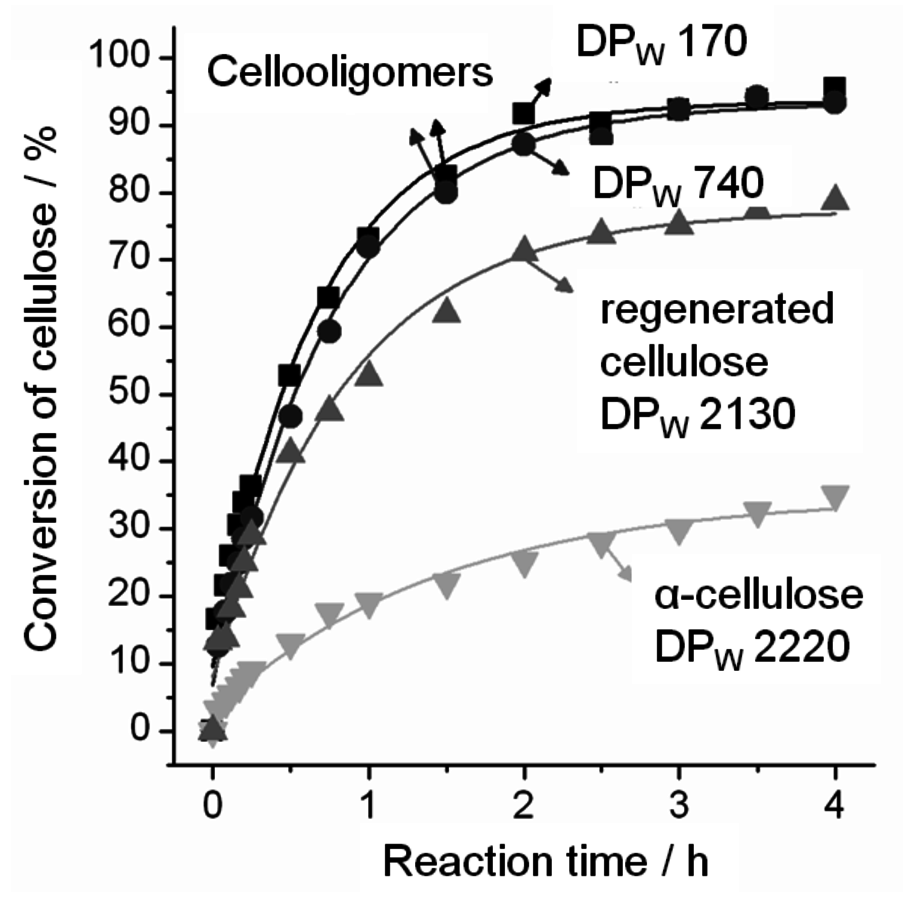
Fig. 13.12: Enzymatic hydrolysis of α-cellulose, cellulose regenerated from IL and cellooligomers. Conditions: substrate (equals 1 g of dry cellulose), cellulase (Celluclast®, 350 U/g substrate), pH 4.5 (acetate buffer), 45 °C [44]
13.5 Final remarks
The biorefining of lignocellulose to produce bio-based chemical assets, and ultimately biofuels, requires solvents. This is the main feature that distinguishes biorefinery processes from the ones currently in practice in oil refineries and petrochemical industries. In the crude oil based industries commonly no solvent is required, because the raw materials and the products are often liquids.
The experience gained in the heterogeneous hydrolysis of cellulose, such as in the two-stage dilute sulfuric acid process [11], shows that a considerable volume of solvent is necessary. For instance, processing 1 kg of dried oven wood uses about 3 L of water. Moreover, for each liter of ethanol, 32 L of industrial waste- water is generated in its production from wood. In a large industrial plant with a capacity to process 10,000 tons of lignocellulosic material a day (wood, straw and others), producing 870 tons of ethanol, the amount of industrial wastewater generated would reach 32,070,000 liters! This large volume of water is enough to supply a town with, for example, 300,000 inhabitants in an industrialized area daily.
Although water is the most environmentally friendly solvent known, this statement is only valid when water is returned clean to the environment. In the case of the dilute acid process, several dehydration products, such as 5-HMF, furfural, formic acid and even some phenols are formed [2]. The concentration of some of these impurities has to be at the ppm level for the proper reuse of water. Accordingly, high-cost advanced oxidation processes for the industrial wastewater cleaning are mandatory.
Looking at the present technologies in development, avoiding the use of solvents in the biorefinery process chains appears to be unfeasible. Accordingly, the use of solvents should be rationally applied. For instance, in the two-stage dilute sulfuric acid process, water functions merely as a dispersant of the substrate in the medium. Replacing water with a solvent that at least swells the lignocellulosic matrix could already improve markedly the performance of the acid-catalyzed hydrolysis of cellulose (and hemicellulose) at lower temperatures than currently in use. However, the best solution would be to perform the process under homogeneous conditions. This would then take advantage of carrying out the hydrolytic process at room temperature, making the overall energetic demands lower.
Although the dissolution of cellulose is a process still not fully comprehended, it is clear from the thermodynamic prediction models that solvents for cellulose are liquids (or solutions) that possess high cohesion energy densities, i.e., are commonly compounds that have high boiling points. Hence, research on advanced processes for solvent reclamation is necessary to avoid energy intensive processes in their reuse.
The use of solvents is a central, but often neglected problem in lignocellulosic biorefining. This most urgently needs to change in order to place cellulosic ethanol, and ultimately the third generation biofuels, as a cost competitive and more sustainable alternative to fossil fuels.
Bibliography
[1] D. Klemm, B. Heublein, B. H., Fink B.. Cellulose: Fascinating Biopolymer and Sustainable Raw Material Angewandte Chemie International Edition 44(22): 76-81 (2007): 76-81.
[2] R. Rinaldi, F. Schüth. Acid Hydrolysis of Cellulose as the Entry Point into Biorefinery Schemes ChemSusChem 2(12): 1096-1107 (2009): 1096-1107.
[3] Y. Nishiyama, J. Sugiyama, J. S., Chanzy J.. Crystal Structure and Hydrogen Bonding System in Cellulose Iα from Synchrotron X-Ray and Neutron Fiber Diffraction Journal of the American Chemical Society 125(47): 14300-14306 (2003): 14300-14306.
[4] A.C. O ' Sullivan. Cellulose: The Structure Slowly Unravels Cellulose 4(3): 173-207 (1997): 173-207.
[5] R. Rinaldi, F. Schüth. Design of Solid Catalysts for the Conversion of Biomass Energy & Environmental Science 2: 610-626 (2009): 610-626.
[6] R. Wolfenden, M.J. Snider. The Depth of Chemical Time and the Power of Enzymes as Catalysts Accounts of Chemical Research 34(12): 938-945 (2001): 938-945.
[7] H. Zhao, J.H. Kwak, J. K., Wang J.H., W. J., Y. Wang. Effects of Crystallinity on Dilute Acid Hydrolysis of Cellulose by Cellulose Ball-Milling Study Energy and Fuels 20(2): 807-811 (2006): 807-811.
[8] P. Kumar, D.M. Barrett, D. B., Delwiche D.M.. Methods for Pretreatment of Lignocellulosic Biomass for Efficient Hydrolysis and Biofuel Production Industrial and Engineering Chemistry Research 48(8): 3713-3729 (2009): 3713-3729.
[9] B. Yang, C.E. Wyman. Pretreatment: The Key to Unlocking Low-Cost Cellulosic Ethanol Biofuels, Bioproducts and Biorefining 2(1): 26-40 (2008): 26-40.
[10] T. Liebert. Cellulose Solvents—Remarkable History, Bright Future., 2010
[11] J.F. Harris, A.J. Baker, A. B., Conner A.J., C. A., A.H. Conner, A. C., Jeffries A.H., J. A., T.W. Jeffries () Two-Stage, Dilute Sulfuric Acid Hydrolysis of Wood: An Investigation of Fundamentals.
[12] T. Shen, P. Langan, P. L., French P., F. P.. Conformational Flexibility of Soluble Cellulose Oligomers: Chain Length and Temperature Dependence Journal of the American Chemical Society 131(41): 14786-14794 (2009): 14786-14794.
[13] N. Huu-Phuoc, H. Nam-Tran, H. NT., Buchmann H.. Experimentally Optimized Determination of the Partial and Total Cohesion Parameters of an Insoluble Polymer (Microcrystalline Cellulose) by Gas-Solid Chromatography International Journal of Pharmaceutics 34(3): 217-223 (1987): 217-223.
[14] R.J. Roberts, R.C. Rowe. The Solubility Parameter and Fractional Polarity of Microcrystalline Cellulose as Determined by Mechanical Measurement International Journal of Pharmaceutics 99(2-3): 157-164 (1993): 157-164.
[15] C.M. Hansen, A. Bjoerkman. The Ultrastructure of Wood from a Solubility Parameter Point of View Holzforschung 52(4): 335-344 (1998): 335-344.
[16] A.F.M. Barton. Handbook of Solubility Parameters., 1983, pp. 153–157
[17] C.M. Hansen. Hansen Solubility Parameters. .
[18] M. Mora-Pale, L. Meli, L. M., Doherty L., D. L.. Room Temperature Ionic Liquids as Emerging Solvents for the Pretreatment of Lignocellulosic Biomass Biotechnology and Bioengineering 108(6): 1229-1245 (2011): 1229-1245.
[19] S. Fischer, H. Leipner, H. L., Thümmler H., T. H.. Inorganic Molten Salts as Solvents for Cellulose Cellulose 10(3): 227-236 (2003): 227-236.
[20] T. Heinze, A. Koschella. Solvents Applied in the Field of Cellulose Chemistry: A Mini Review Polimeros 15(2): 84-90 (2005): 84-90.
[21] A.F. Turbak, R.B. Hammer, R. H., Davies R.B.. Cellulose Solvents Chemtech 10(1): 51-57 (1980): 51-57.
[22] A. Pinkert, K.N. Marsh, K. M., Pang K.N.. Ionic Liquids and Their Interaction with Cellulose Chemical Reviews 109: 6712-6728 (2009): 6712-6728.
[23] N. Sun, H. Rodriguez, H. R., Rahman H.. Where are Ionic Liquid Strategies most Suited in the Pursuit of Chemicals and Energy from Lignocellulosic Biomass? Chemical Communications 47: 1405-1421 (2011): 1405-1421.
[24] M. Zavrel, D. Bross, D. B., Funke D., F. D.. High-Throughput Screening for Ionic Liquids Dissolving (Ligno-)Cellulose Bioresource Technology 9(100): 2580-2587 (2009): 2580-2587.
[25] J. Kahlen, K. Masuch, K. M.. Modelling cellulose solubilities in ionic liquids using COSMO-RS Green Chemistry 12: 2172-2181 (2010): 2172-2181.
[26] R.P. Swatloski, S.K. Spear, S. S., Holbrey S.K.. Dissolution of Cellulose with Ionic Liquids Journal of the American Chemical Society 124(18): 4974-4975 (2002): 4974-4975.
[27] N. Meine, F. Benedito, F. B.. Thermal Stability of Ionic Liquids Assessed by Potentiometric Titration Green Chemistry 12: 1711-1714 (2010): 1711-1714.
[28] P.J. Scammells, J.L. Scott, J. S.. Ionic Liquids: The Neglected Issues Australian Journal of Chemistry, 58(3): 155-169 (2005): 155-169.
[29] R. Rinaldi. Instantaneous Dissolution of Cellulose in Organic Electrolyte Solutions Chemical Communications 47: 511-513 (2011): 511-513.
[30] A.W.T. King, J. Asikkala, J. A., Mutikainen J., M. J.. Distillable Acid-Base Conjugate Ionic Liquids for Cellulose Dissolution and Processing Angewandte Chemie International Edition 50(28): 76-81 (2011): 76-81.
[31] F. Bergius. Conversion of Wood to Carbohydrates Industrial and Engineering Chemistry 29(3): 247-253 (1937): 247-253.
[32] M.A. Harmer, A. Fan, A. F., Liauw A.. A New Route to High Yield Sugars from Biomass: Phosphoric-Sulfuric Acid Chemical Communications (2009): 6610-6612.
[33] P.L. Ragg, P.R. Fields, P. F.. The Development of a Process for the Hydrolysis of Lignocellulosic Waste Philosophical Transactions of the Royal Society 321: 537-547 (1987): 537-547.
[34] S. Wei, V. Kumar, V. K.. Phosphoric Acid Mediated Depolymerization and Decrystallization of Cellulose: Preparation of Low Crystallinity Cellulose - A New Pharmaceutical Excipient International Journal of Pharmaceutics 142(2): 175-181 (1996): 175-181.
[35] R.M. Almeida, Li de, L. d., J. Li, J. L., Nederlof J.. Cellulose Conversion to Isosorbide in Molten Salt Hydrate Media ChemSusChem 3(3): 325-328 (2010): 325-328.
[36] N.J. Cao, Q. Xu, Q. X.. Acid Hydrolysis of Cellulose in Zinc Chloride Solution Applied Biochemistry and Biotechnology 51(1): 21-28 (1995): 21-28.
[37] J.F. Saeman, J.L. Bubl, J. B.. Quantitative Saccharification of Wood and Cellulose Industrial Engineering Chemistry, Analytical Edition 17(1): 35-37 (1945): 35-37.
[38] M.R. Ladisch. Fermentable Sugars from Cellulosic Residues Process Biochemistry 14(1): 21-25 (1979): 21-25.
[39] S.V. Vyver, Geboers de, G. d., J. Geboers. Recent Advances in the Catalytic Conversion of Cellulose ChemCatChem 3(1): 82-94 (2011): 82-94.
[40] C. Li, Z.K. Zhao. Efficient Acid-Catalyzed Hydrolysis of Cellulose in Ionic Liquid Advanced Synthesis Catalysis 349(11-12): 1847-1850 (2007): 1847-1850.
[41] J.B. Binder, R.T. Raines. Fermentable Sugars by Chemical Hydrolysis of Biomass Proceedings of the National Academy of Sciences of the United States of America 107(10): 4516-4521 (2010): 4516-4521.
[42] R. Rinaldi, N. Meine, N. M., Stein N., S. N.. Which Controls the Depolymerization of Cellulose in Ionic Liquids: The Solid Acid Catalyst or Cellulose? ChemSusChem 3(2): 266-276 (2010): 266-276.
[43] R. Rinaldi, R. Palkovits, R. P.. Depolymerization of Cellulose Using Solid Catalysts in Ionic Liquids Angewandte Chemie International Edition 47(42): 8047-8050 (2008): 8047-8050.
[44] R. Rinaldi, P. Engel, P. E., Buechs P., B. P.. An Integrated Catalytic Approach to Fermentable Sugars from Cellulose ChemSusChem 3(10): 1151-1153 (2010): 1151-1153.
Footnotes
The term cellulose I describes two polymorphs, Iα and Iβ. The polymorph Iα is found in the cell wall of some algae and in bacterial cellulose, whereas the polymorph Iβ is predominant in cotton, wood, and ramie fibers. However, both polymorphs can be found in the same sample and along the same microfibril. This made the first investigations of the crystalline structure of Cellulose I cumbersome and controversial [3,4].