12.1 Introduction
Individual mobility together with automotive cargo transportation represents one of the fundamental incentives for economic stability and growth. However, the energy supply within the transport sector is currently facing major changes due to the urgent need to accomplish increasing independence from fossil raw materials and to reduce the associated carbon dioxide emission. Scenarios based on these assumptions predict an increasing technological diversification of the transportation sector including the use of various alternative energy carriers, such as natural gas, ethanol, or hydrogen, and the application of electrically powered automobiles. All possible scenarios have in common that electrically powered vehicles are expected to impact mostly on urban transportation, whereas internal combustion engines will remain the prevailing propulsion systems for long-distance and heavy-duty applications. Due to their superior energy density as well as proven storage and distribution concepts, liquid chemical compounds represent the energy carrier of choice for these applications.
Projections of the International Energy Agency (IEA) predict that the use of fuels from biomass will continuously increase, providing up to 27% of all transportation fuels in 2050, compared to only 3% today. The competition to the food supply chain as well as the critical energy and
 -balance have triggered intensive research on sustainable biofuel production beyond the established first generation products based on sugarcane, corn, maize or palm oil. These established processes only use certain parts of the plant material for fuel production. With the exception of specific regional situations such as sugarcane in Brasil, they therefore imply severe limitations on the way to a sustainable long-term solution. Significant progress has been achieved on large-scale implementation of second generation biofuels, which intend to use the non-food relevant part of the plant material. One prominent example is the Biomass-to-Liquid (BtL) approach, where biomass materials are converted to syngas and subsequently transformed to alkane mixtures. In mid-term, second generation biofuels are expected to contribute up to 4% of all utilized fuels in Europe in 2020. However, current efforts are aiming mainly at the substitution of fossil fuels based on hydrocarbon mixtures without capitalizing on the potential of innovative fuel design and improved engine systems. In order to fully exploit the engine potential in terms of emissions and fuel efficiency, the ideal properties of future fuels may differ considerably from those of current conventional fuels, making it attractive to concomitantly tailor the combustion systems to the fuel properties and vice versa.
-balance have triggered intensive research on sustainable biofuel production beyond the established first generation products based on sugarcane, corn, maize or palm oil. These established processes only use certain parts of the plant material for fuel production. With the exception of specific regional situations such as sugarcane in Brasil, they therefore imply severe limitations on the way to a sustainable long-term solution. Significant progress has been achieved on large-scale implementation of second generation biofuels, which intend to use the non-food relevant part of the plant material. One prominent example is the Biomass-to-Liquid (BtL) approach, where biomass materials are converted to syngas and subsequently transformed to alkane mixtures. In mid-term, second generation biofuels are expected to contribute up to 4% of all utilized fuels in Europe in 2020. However, current efforts are aiming mainly at the substitution of fossil fuels based on hydrocarbon mixtures without capitalizing on the potential of innovative fuel design and improved engine systems. In order to fully exploit the engine potential in terms of emissions and fuel efficiency, the ideal properties of future fuels may differ considerably from those of current conventional fuels, making it attractive to concomitantly tailor the combustion systems to the fuel properties and vice versa.
The Cluster of Excellence Tailor-Made Fuels from Biomass at RWTH Aachen University addresses the fundamental scientific and technological challenges associated with this approach toward next generation biofuels.1 Researchers from RWTH Aachen and the Max-Planck-Institut für Kohlenforschung have formed an interdisciplinary, integrated network comprising scientific expertise in the fields of chemistry, biology, reaction and process engineering, and combustion engineering. Complementary to current state-of-the-art biofuel concepts, the Cluster’s research program aims at the selective (bio-)chemical conversion of lignocellulosic biomass into molecularly defined fuel compounds that are not merely viewed as substitutes for existing products, but recognized as an opportunity to improve the performance of internal combustion engines.
12.2 Conceptual Approach to Tailor-Made Fuels via Combined Product and Process Design
The initial step in developing production pathways for the desired new fuel candidates is to define target molecules comprising the envisaged physical properties, which are beneficial for the application in internal combustion engines. Based on these novel fuel molecules, potential production pathways have to be identified. For this task we have proposed to apply a design strategy in analogy to the retrosynthetic approach commonly used in organic chemistry [1]. In such an analysis, the targeted molecules are traced back to the starting materials step by step through building blocks and intermediates that are readily available (Figure 12.1). In organic synthesis based on the current petrochemical supply chain, the retrosynthetic analysis typically reduces the complexity and functionality of the products on the way to the starting materials. Synthetic methods for the formation of carbon-carbon and carbon-heteroatom bonds connect the simpler building blocks with the more complex product structures. In contrast, the target products of the biomass-based supply chain are typically less oxidized and of lower molecular weight than the biopolymer feedstocks. Therefore, new catalytic methods for selective de- and re-functionalization (mainly deoxygenation) in liquid phase have to be developed to connect the structures along the possible pathways identified in the retrosynthetic analysis.
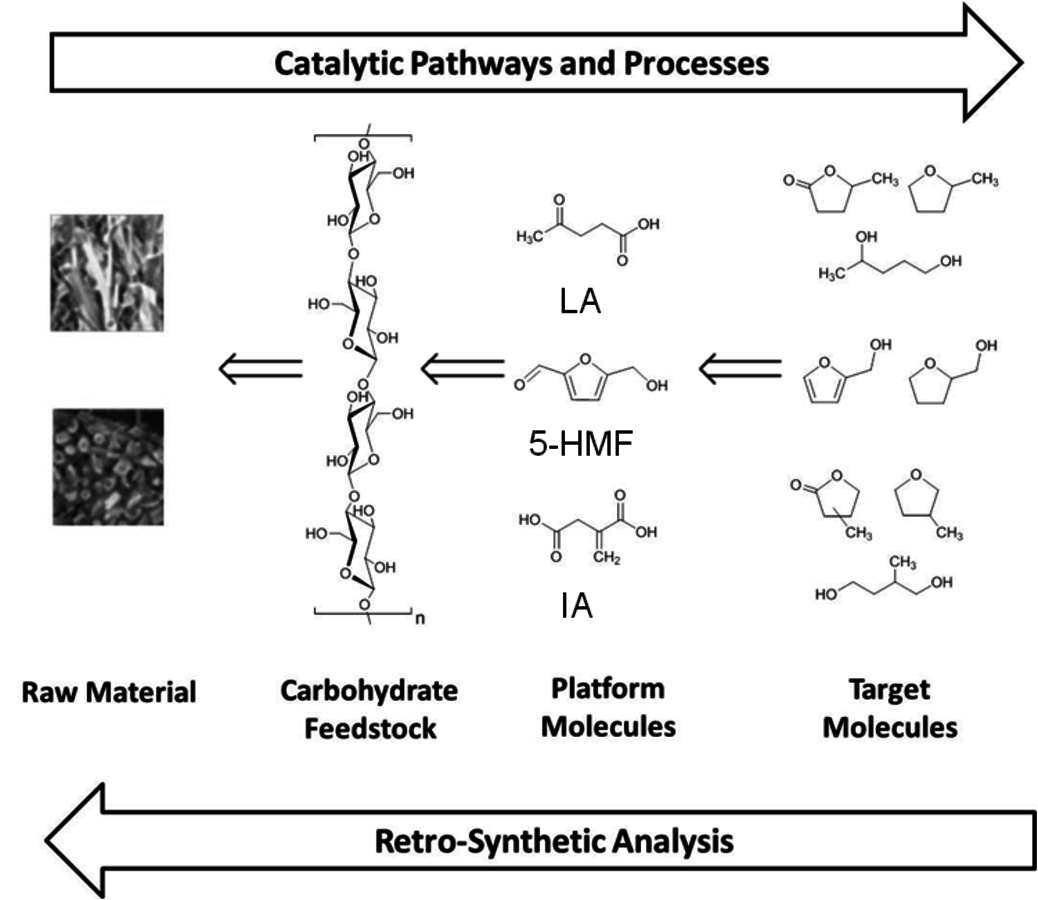
Fig. 12.1: Concept for the targeted development of new routes to potential fuel compounds based on lignocellulose as raw material
Figure 12.1 exemplifies this approach for cyclic esters, ethers and furan derivatives as products. Based on the literature background [2], these molecular structures have been identified as promising targets for potential fuel components and continue to be refined further through the interdisciplinary collaboration within the Cluster. The specific structures can be traced back to well-defined intermediates, levulinic acid (LA), itaconic acid (IA) and 5-hydroxymethylfurfural (5-HMF), which have been selected from the range of so-called platform chemicals available from the carbohydrate feedstock of biomass. Finally, the required cellulose and hemicellulose fractions can be obtained by the disintegration of the diverse lignocellulosic raw material coupled with selective separation processes. The following sections will describe some selected recent developments and advances for these steps to highlight the challenges and opportunities for catalysis research resulting from this targeted and rational approach to next generation biofuels and biomass-derived chemicals.
12.3 From Intermediates to Products
12.3.1 Chemicals and Fuels from the Platform Chemicals Levulinic and Itaconic Acid
2-Methyltetrahydrofurane (2-MTHF) and γ-valerolactone (GVL) can be obtained from levulinic acid (LA), whereas the isomeric compounds 3-MTHF and methyl-butyrolactone (MBL) are accessible from itaconic acid (IA). Even for these seemingly direct conversions, a large number of pathways are possible. For example, as many as nineteen different connections are conceivable to lead from IA to 3-MTHF. Researchers within the Cluster have developed a mathematical model for the reaction network flux analysis that allows characterization of such competing pathways according to criteria such as yield, mass-, or energy efficiency [3]. Thus, the possible solutions can be ranked accordingly, and the most desirable pathways can be selected a priori as primary goals for the catalysis research.
A prerequisite to enable these sustainable pathways from platform molecules to tailored fuel candidates represents the development of flexible catalytic systems. The envisaged systems for the highly selective defunctionalisation can be based on catalytic dehydration-hydrogenation, decarboxylation, decarbonylation, or dehydroxylation strategies. For example, the defunctionalization pathway of LA to 2-MTHF, that was selected according to the criteria defined above, comprises a series of consecutive hydrogenation and dehydration steps. Therefore, it requires a multifunctional catalytic system, which was not available at the beginning of the study [1]. Thus, the conceptually combined analysis of the energetic balance and the required molecular transformations allows identification of defined challenges for catalyst development.
The sequence of molecular transformations from LA via γ-valerolactone (GVL), to 1,4-pentanediol (1,4-PDO), and finally 2-MTHF is shown in the scheme in Figure 12.2. The stepwise process consists of a consecutive series of hydrogenations (green) and dehydrations (blue) via well-defined, but short-lived intermediates (in brackets). Initially, the keto function of LA is reduced to γ-hydroxy carboxylic acid, and spontaneous intramolecular esterification yields γ-valerolactone (GVL). The reduction of the C=O bond gives a hemiacetal, which is in equilibrium with the open hydroxy aldehyde. A further hydrogenation step then forms 1,4-pentane-diol (1,4-PDO). Finally, acid catalyzed intramolecular etherification yields 2-MTHF.
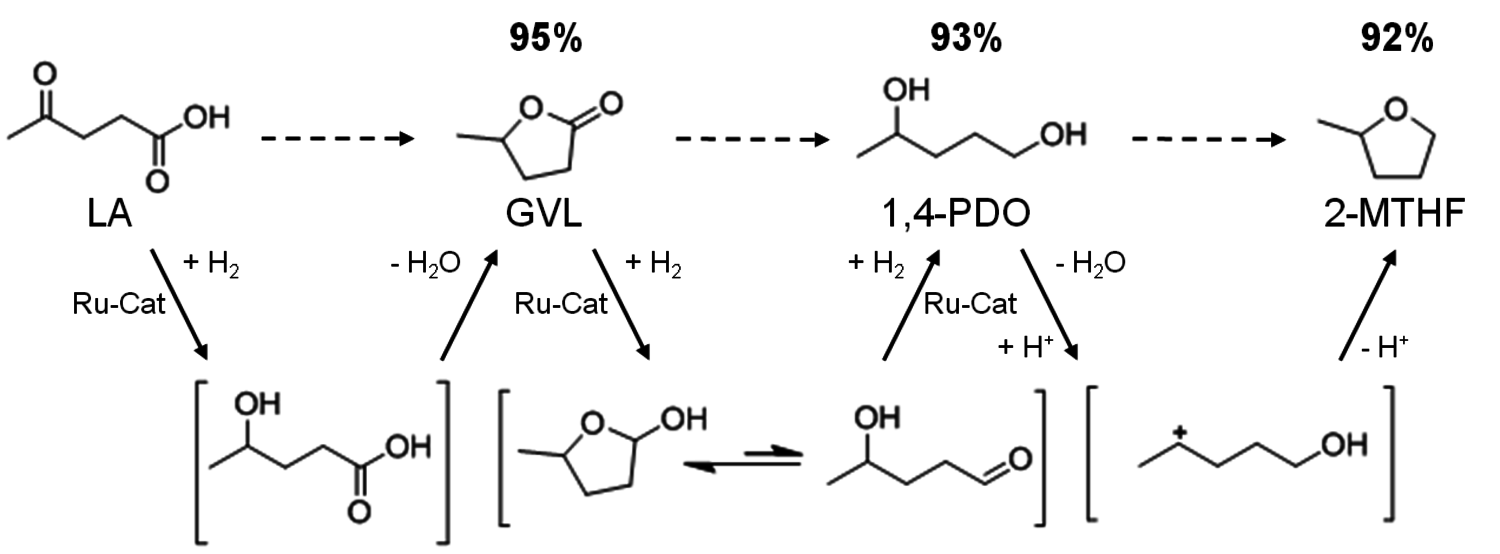
Fig. 12.2: Reaction sequence and maximum yields for the selective conversion of levulinic acid (LA) into γ-valerolactone (GVL), 1,4-pentanediol (1,4-PDO), and 2-methyltetrahydrofuran (2-MTHF)
For the initial hydrogenation step of LA to GVL, heterogeneous systems were applied previously with great success and ruthenium catalysts supported on carbon showed favorable selectivity [4]. Recently, the combination of ruthenium on silica and supercritical
 as reaction and extraction media even enabled the continuous production of GVL from LA [5]. Horvath and co-workers were able to apply a homogeneous ruthenium system based on Ru(acac)3 and a monodentate phosphine ligand for this transformation. This system promoted quantitative conversion of LA to GVL after 8 hours at a moderate reaction temperature of 135 °C [6]. Based on these pioneering studies [6,7], a multifunctional system based on the ruthenium precursor Ru(acac)3, a phosphine ligand, and suitable additives were developed. As LA has a melting point of 31–33 °C and a high thermal stability, it provides a suitable reaction medium for the homogeneously catalyzed processes without the need of the addition of any external solvent. The key feature of this new system represents the tuneable product selectivity by control of the hydrogenation activity through the choice of ligand combined with adjustment of the acidity of the reaction medium through the additives (Figure 12.3). With the monodentate ligand trioctylphosphine P(n-Oct)3, GVL is formed in almost quantitative yield in agreement with literature data [6-8]. The use of the tripodal phosphine ligand 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos) leads to a significantly more active hydrogenation catalyst that reduces the cyclic ester further. In absence of additives, 1,4-PDO is formed in 95% yield. The addition of the acidic ionic liquid (4-sulfobutyl)imidazolium-p-toluenesulfonate shifts the hydrogenation beyond the diol stage giving 2-MTHF in 87% yield. This could be even further improved to 92% yield by using additional stabilization by NH4PF6 in catalytic amounts. With this modular catalyst system, three valuable products are accessible in excellent yields from one substrate, emphasizing the potential of the selective defunctionalization approach. The scheme in Figure 12.2 summarizes the optimum yields for the direct conversion of LA and the remarkable control and flexibility offered by the multifunctional molecular catalyst system.
as reaction and extraction media even enabled the continuous production of GVL from LA [5]. Horvath and co-workers were able to apply a homogeneous ruthenium system based on Ru(acac)3 and a monodentate phosphine ligand for this transformation. This system promoted quantitative conversion of LA to GVL after 8 hours at a moderate reaction temperature of 135 °C [6]. Based on these pioneering studies [6,7], a multifunctional system based on the ruthenium precursor Ru(acac)3, a phosphine ligand, and suitable additives were developed. As LA has a melting point of 31–33 °C and a high thermal stability, it provides a suitable reaction medium for the homogeneously catalyzed processes without the need of the addition of any external solvent. The key feature of this new system represents the tuneable product selectivity by control of the hydrogenation activity through the choice of ligand combined with adjustment of the acidity of the reaction medium through the additives (Figure 12.3). With the monodentate ligand trioctylphosphine P(n-Oct)3, GVL is formed in almost quantitative yield in agreement with literature data [6-8]. The use of the tripodal phosphine ligand 1,1,1-tris(diphenylphosphinomethyl)ethane (triphos) leads to a significantly more active hydrogenation catalyst that reduces the cyclic ester further. In absence of additives, 1,4-PDO is formed in 95% yield. The addition of the acidic ionic liquid (4-sulfobutyl)imidazolium-p-toluenesulfonate shifts the hydrogenation beyond the diol stage giving 2-MTHF in 87% yield. This could be even further improved to 92% yield by using additional stabilization by NH4PF6 in catalytic amounts. With this modular catalyst system, three valuable products are accessible in excellent yields from one substrate, emphasizing the potential of the selective defunctionalization approach. The scheme in Figure 12.2 summarizes the optimum yields for the direct conversion of LA and the remarkable control and flexibility offered by the multifunctional molecular catalyst system.
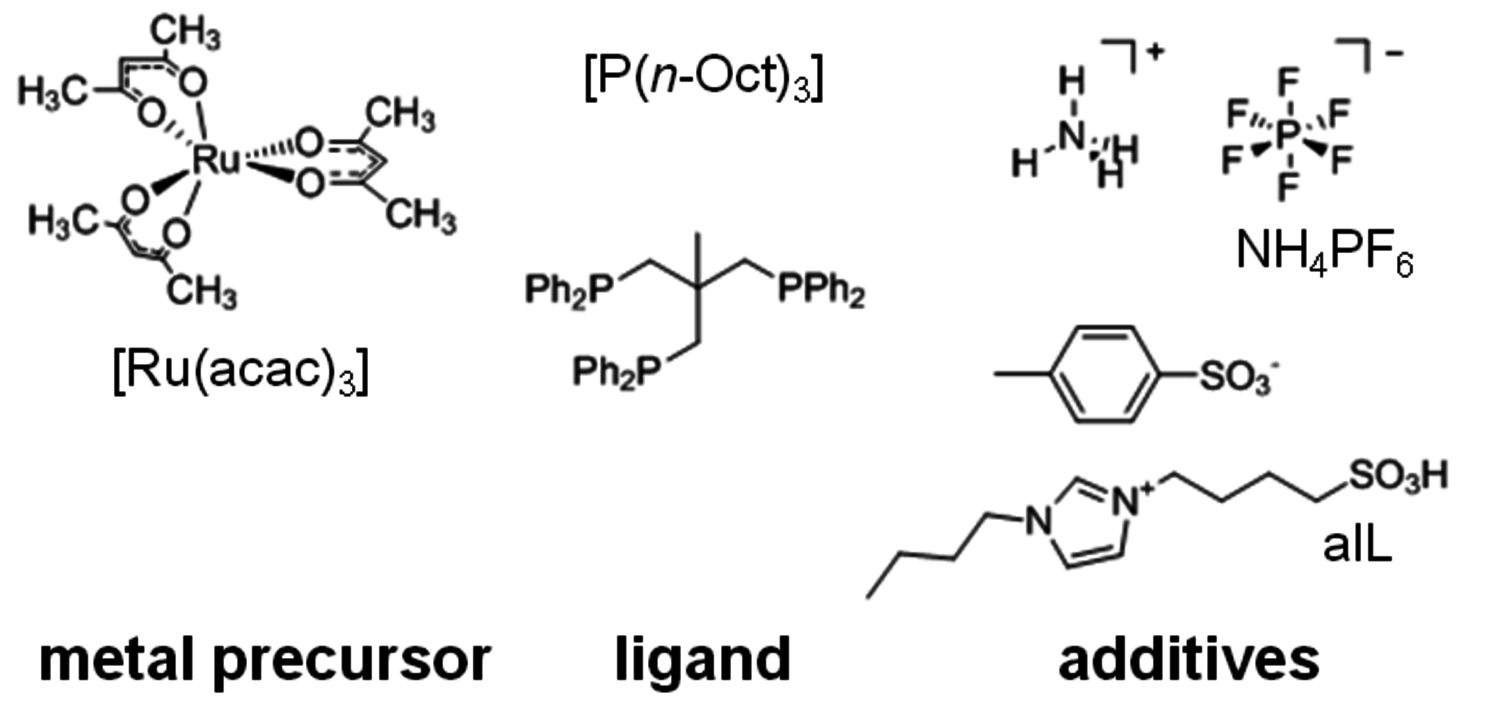
Fig. 12.3: Components for the generation of the multifunctional catalyst. aIL: acidic ionic liquid
While 1,4-PDO is primarily interesting for applications in the polymer industry [9], GVL and 2-MTHF are discussed as promising fuel candidates. GVL has been employed as additive in diesel and gasoline fuels with a RON of 96 [7]. 2-MTHF has a lower oxygen content as well as a higher combustion enthalpy and therefore should exhibit beneficial engine properties. In fact, a blend of 70% 2-MTHF and 30% dibutylether showed promising results in diesel engine tests. In comparison to standard EN595 diesel, the oxygenate blend allowed a nearly soot-free combustion even at high engine loads. Furthermore, hazardous
 emissions could be significantly reduced due to the low internal combustion temperature [10]. Previously, 2-MTHF has been already approved by the U.S. Department of Energy as an additive in gasoline blends which consist of 35% liquids from natural gas and 45% ethanol. These P Type series fuels exhibit octane numbers ranging from 89 to 93.
emissions could be significantly reduced due to the low internal combustion temperature [10]. Previously, 2-MTHF has been already approved by the U.S. Department of Energy as an additive in gasoline blends which consist of 35% liquids from natural gas and 45% ethanol. These P Type series fuels exhibit octane numbers ranging from 89 to 93.
The ruthenium/triphos catalyst system, which efficiently promoted the conversion of the C6-sugar derived platform molecule LA, could also be readily adapted to the intermediate itaconic acid (IA), which can be assessed at least in principle by fementation routes from C6 or C5-sugars. This substrate cannot be used for neat reactions, but bio-based MTHF could be used as a reaction solvent. The detailed transformation sequence is shown in Figure 12.4. In analogy to the LA reduction, the transformation of IA to 3-methyltetrahydrofuran (3-MTHF) involves a series of hydrogenation and dehydration steps. The hydrogenation of the C=C bond in IA to form methyl succinic acid (MSA) is straightforward with ruthenium phosphine catalysts, but the reduction of the free carboxylic acid function with molecular hydrogen is very challenging for a homogeneous organometallic catalyst. Reduction of one carboxyl group and subsequent intra molecular esterification yields 2- and 3-Methyl-γ-butyrolacton (3- and 2-MGBL). At 180 °C selective formation of the two isomeric lactones was obtained in a combined yield of about 80% with the Ru/triphos catalyst in the absence of any additives. Once this lactone stage is reached, further conversion into 2-methyl-butanediol (2-MBDO) and the tetrahydrofuran derivative 3-MTHF is achieved by increasing the temperature above 195 °C and can be controlled as for LA through the choice of additives. The diol 2-MBDO could be obtained in excellent yield of up to 93% and the combination of p-toluenesulfonic acid (p-TsOH) and
 as additive afforded 3-MTHF in 95% yield.
as additive afforded 3-MTHF in 95% yield.
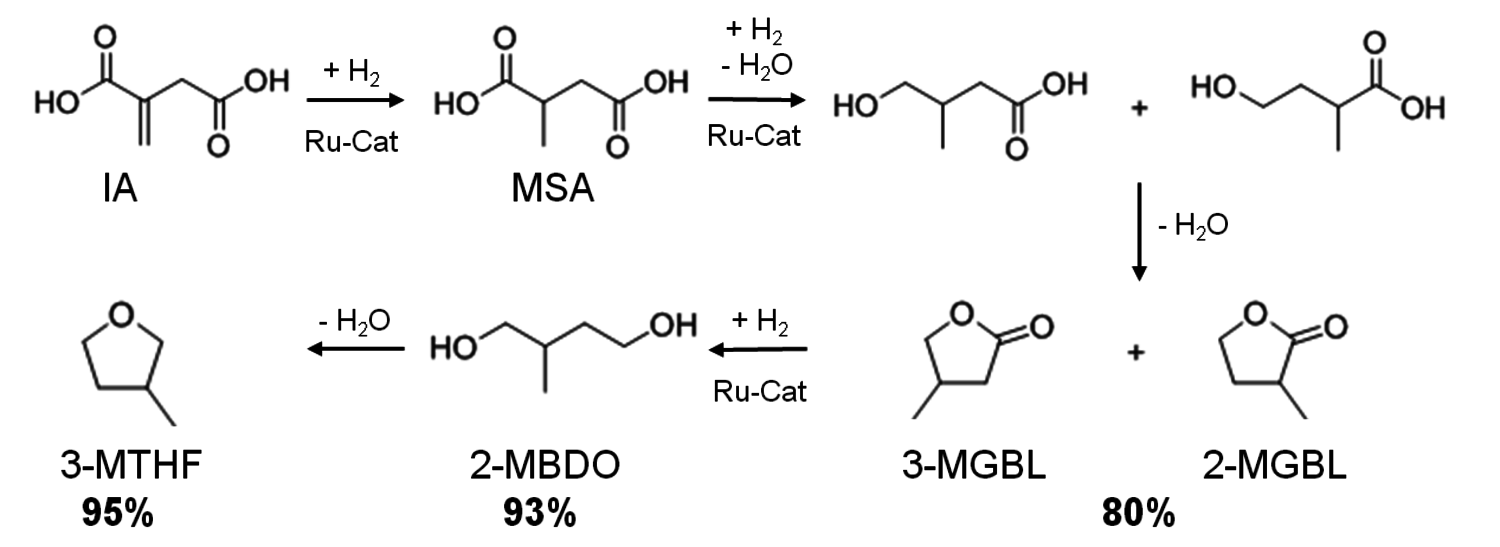
Fig. 12.4: Reaction sequence and maximum yields for the selective conversion of itaconic acid (IA) into lactones (MGBL), 2-methylbutanediol (2-MBDO), and 3-methyltetrahydrofuran (3-MTHF)
In order to gain insight into the mechanistic details of the catalytic cycle, a density functional theory (DFT) study was performed and corroborated with experimental data from catalytic processes and NMR investigations. For the cationic fragment [Ru(triphos)H]+ as catalytically active unit, a common mechanistic pathway for the reduction of the C=O functionalities in the various substrates could be identified (Figure 12.5) [11]. The reduction results from hydride transfer on to the carbonyl or carboxyl carbon via transition states typical for migratory insertion. The subsequent hydrogenolysis of the metal-oxide unit involves proton transfer via σ-bond metathesis from a coordinated dihydrogen molecule, regenerating at the same time the catalytically active Ru-H unit. The interplay between the classical and the non-classical metal hydride coordination provides an overall pathway that does not require changes in the formal oxidation state of the ruthenium center. The energetic spans for the reduction of the different functional groups increase in the order aldehyde < ketone < lactone
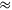 carboxylic acid. Although other mechanistic principles cannot be excluded at this stage, this reactivity pattern as well as the absolute values for the energy barriers are in full agreement with experimentally observed activities and selectivities, forming a rational basis for further catalyst development.
carboxylic acid. Although other mechanistic principles cannot be excluded at this stage, this reactivity pattern as well as the absolute values for the energy barriers are in full agreement with experimentally observed activities and selectivities, forming a rational basis for further catalyst development.
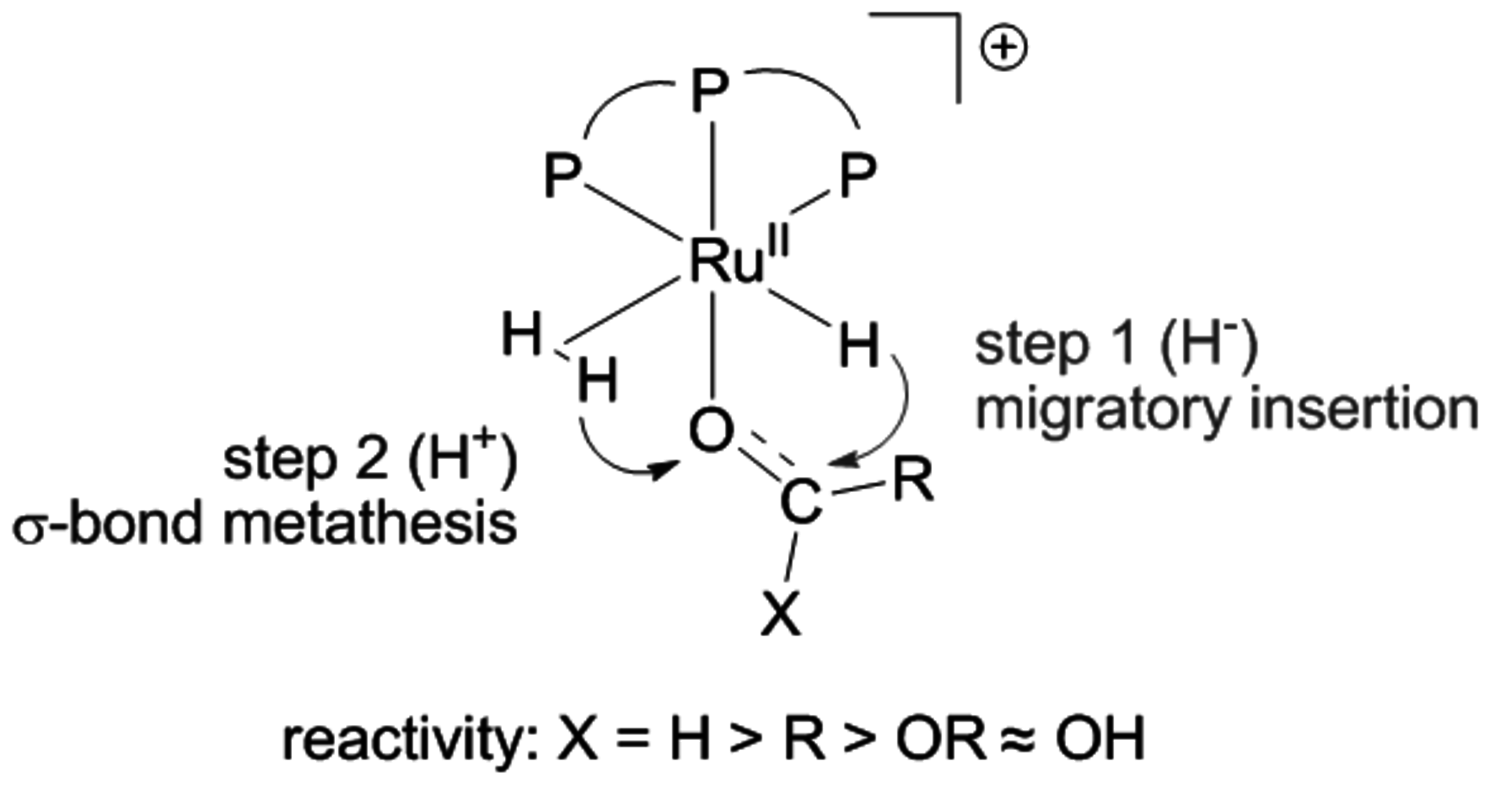
Fig. 12.5: Possible common mechanism for the reduction of carbonyl and carboxyl groups using the Ru-triphos catalytic system
Based on the optimized reaction conditions, a flow sheet for a possible continuous production process for 2-MTH from LA was developed with methods of conceptual process design (Figure 12.6, [1]). In the optimum process scenario, hydrogen and levulinic acid are heated and compressed to reaction conditions and fed into a plug flow reactor. The homogeneous catalyst system is introduced with the liquid substrate in the starting phase of the process. The solvent-free conditions greatly facilitate the isolation and downstream processing of the 2-MTHF product. Under continuous conditions the reactor effluent is vaporized and the remaining liquid stream containing the catalyst and additives is recycled into the reactor. The vapour stream from the flash distillation, which consists of the products 2-MTHF and the water formed from the dehydration steps, is condensed and fed to the decanter of a hetero-azetropic distillation system. The product is recovered in high purity at the bottom of the respective column.
Heat integration between the catalyst recycling and the product recovery can significantly reduce the energy demand of the process, which is in the order of 3% of the energy content of the recovered 2-MTHF. A preliminary economic evaluation revealed that the production costs are dominated mainly by raw materials and catalyst costs. Catalyst productivities corresponding to a turnover number in the order of 105 mol product/mol Ru were estimated to be necessary for the process to become feasible. Although a challenging target, this is well in the range of TON values obtained with transition metal phosphine catalysts in industrial applications [12] or with advanced immobilization concepts [13,14]. It is also important to note that the process does not critically depend on the economy of scale, in contrast to conventional refinery or BTL units. This makes this approach well compatible with the logistics of biomass utilization, which are expected to be more decentralized than current refinery technology due to the large areal distribution of typical lignocellulosic feedstock.
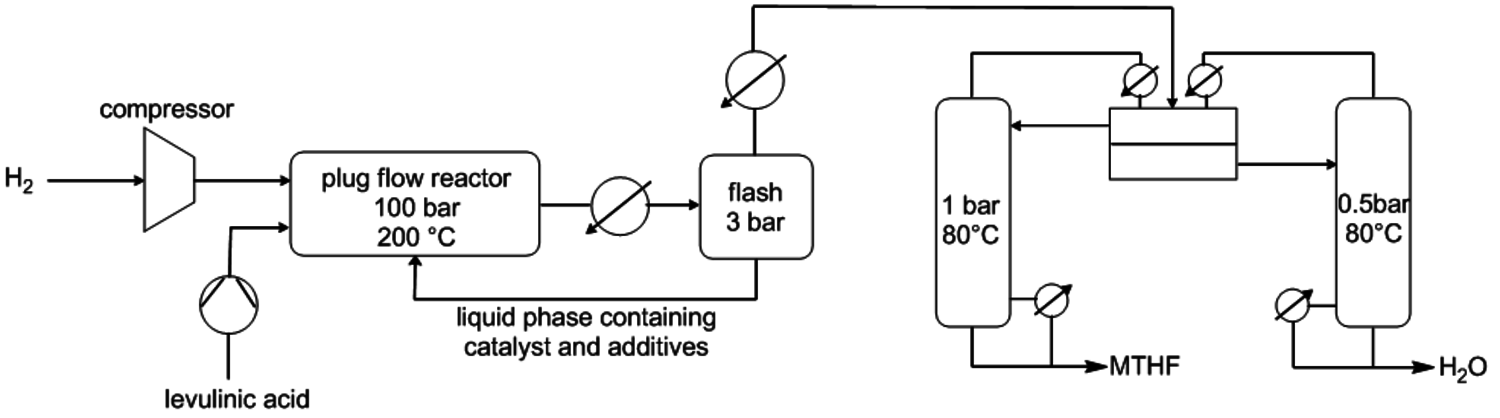
Fig. 12.6: Flow sheet for a possible LA to 2-MTHF process
12.3.2 Chemicals and Fuels from the Platform
Molecules Furfural and 5-HMF
As can be seen in Figure 12.1, further derivatives from tetrahydrofuran or furan structures can be obtained starting from 5-HMF. Iterative progress in the product design suggests that these compounds provide access to further potential fuels, in particular by variation of the carbon number and potential esterification or etherification of the -OH groups [15]. The currently established production route of tetrahydrofurfuryl alcohol (THFA) is based on the hydrogenation of furfural (FF), which is produced on industrial-scale through dehydration of C5-sugars. In light of the larger availability of the C6-sugars over C5-sugars in biomass feedstock, it would be attractive to gain access to THFA and its follow-up products from the C6-sugar platform if fuel applications are envisaged. In this respect the decarbonylation of 5-hydroxymethylfurfural (5-HMF) would represent a viable entry point (Figure 12.7). 5-HMF is accessible from C6-sugars via dehydration processes, albeit the currently employed starting material to obtain high yields is fructose. Due to the high functionalization of 5-HMF, the compound is very reactive and prone to polymerization yielding tar-like humins at higher temperatures. Additionally, the further transformation to the coupled products levulinic acid and formic acid often leads to complex mixtures and only modest yields.
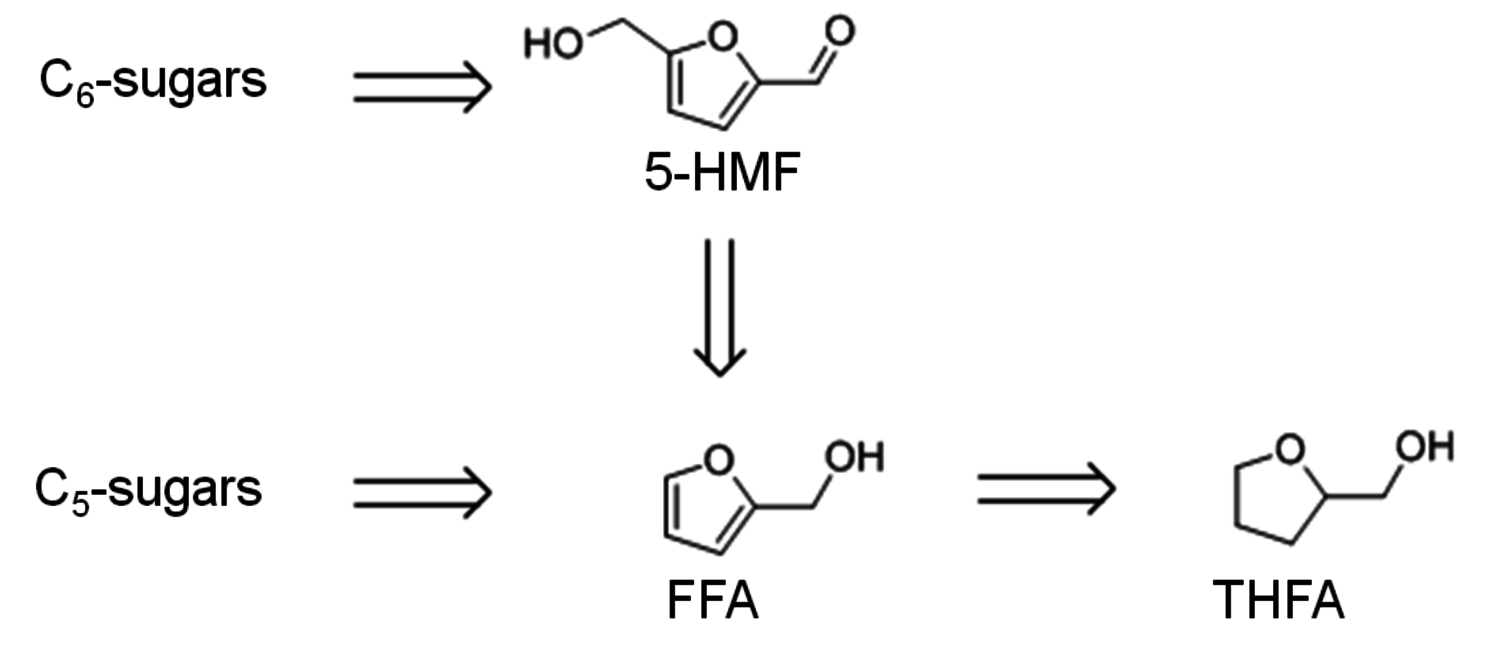
Fig. 12.7: Retrosynthetic connection of the C6 and C5 sugar process chains for furfuryl alcohol (FFA) and tetrahydrofurfuryl alcohol (THFA)
The combination of an efficient iridium-based catalyst in combination with an optimized reaction medium enabled to overcome these limitations at least partly, opening for the first time access to the cyclic C5 alcohols from the C6 feedstock via decarbonylation. The catalyst system was prepared from the iridium precursor
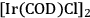 and a monodentate phosphine ligand (tricyclohexylphosphine or trioctylphosphine) [16]. In this decarbonylation reaction the addition of 50 bar
and a monodentate phosphine ligand (tricyclohexylphosphine or trioctylphosphine) [16]. In this decarbonylation reaction the addition of 50 bar
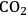 pressure successfully suppressed the undesired polymerization/decomposition processes, most likely by formation of an expanded liquid phase [14]. Thus, full conversion of 5-HMF to FFA could be obtained with 95 % selectivity after 12 hours (Figure 12.8). Beyond the specific target molecule, this result demonstrates that decarbonylation provides a useful route for the reduction of the oxygen content in bio-based supply chains, avoiding the use of high-energy and costly hydrogen equivalents.
pressure successfully suppressed the undesired polymerization/decomposition processes, most likely by formation of an expanded liquid phase [14]. Thus, full conversion of 5-HMF to FFA could be obtained with 95 % selectivity after 12 hours (Figure 12.8). Beyond the specific target molecule, this result demonstrates that decarbonylation provides a useful route for the reduction of the oxygen content in bio-based supply chains, avoiding the use of high-energy and costly hydrogen equivalents.

 pressures
pressuresFig. 12.8: Product mixtures and FFA yields of the decarbonylation after reaction under different
pressures
The resulting furfuryl alcohol (FFA) is readily hydrogenated by ionic liquid stabilized Ru-nanoparticles to obtain tetrahydrofurfuryl alcohol (THFA), yet another target molecule for engine applications. In addition, THFA could also be used as a solvent for the decarbonylation as well as for the hydrogenation reaction, highly facilitating subsequent downstream processing (Figure 12.9). A reasonably high octane number of 83 was reported for both FFA and THFA and detailed properties as fuel additives are consequently under current investigation.
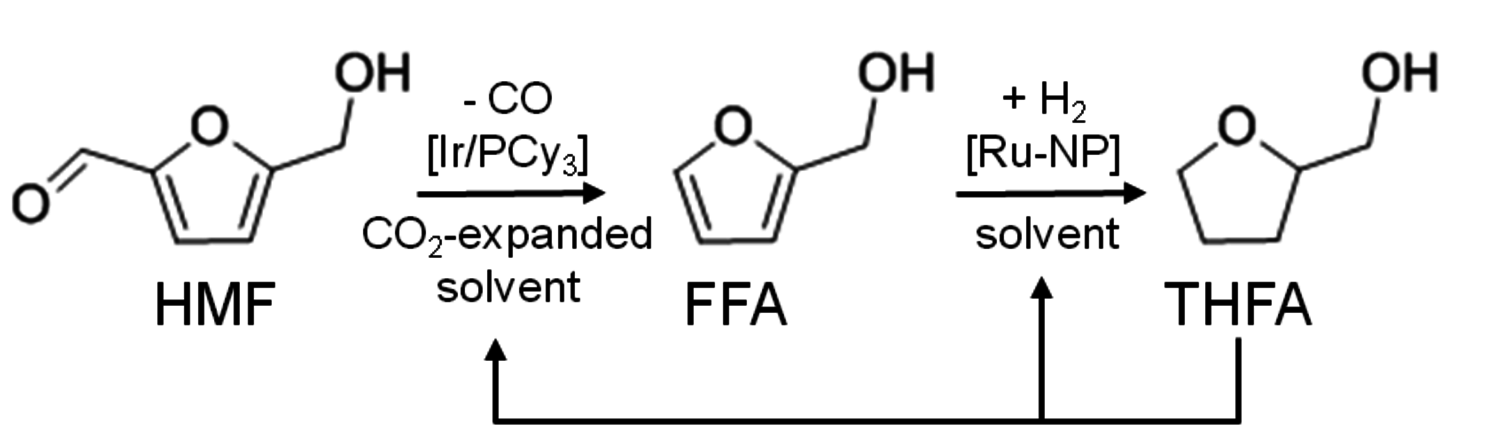
Fig. 12.9: The integrated development and optimization of catalysts and reaction media lead to a highly selective decarbonylation of 5-HMF to FFA with an iridium/phosphine-catalyst in the presence of compressed carbon dioxide. Subsequent hydrogenation over ruthenium nanoparticles leads to THFA, a possible solvent for the decarbonylation reaction.
Another furan-derivative based on 5-HMF, which has shown interesting combustion properties is 2,5 Dimethylfuran (2,5-DMF) [17]. Dumesic and co-workers developed an integrated catalytic system, which enabled the direct conversion of fructose to 2,5-DMF [18]. First, fructose is dehydrated to 5-HMF by HCl in aqueous media, while a second organic layer allows in-situ extraction leading to a high selectivity toward 5-HMF. Once HCl and water are removed by evaporation, this extracting phase is fed directly to a hydrogenolysis reactor. Here, a carbon-supported copper-ruthenium catalyst efficiently promotes the deoxygenation of 5-HMF with four equivalents of hydrogen to afford 2,5-DMF. Finally, the product and water contaminants are evaporated from the reaction solution, which can be recycled back to the dehydration reactor. Since 2,5-DMF is not miscible with water, it separates spontaneously and therefore can be easily isolated. In this way the bio-catalytic formation of fructose, followed by homogeneously acid-catalyzed dehydration and heterogeneously catalyzed deoxygenation of 5-HMF are efficiently coupled into an integrated process concept.
In addition, the key intermediates of the C6 and C5 carbohydrate platform, 5-hydroxymethylfurfural (5-HMF) and furfural, can also be further modified by the incorporation of additional building blocks, for example through aldol-condensa- tion with acetone, leading to compounds with higher carbon numbers (Figure 12.10). Such compounds are promising targets, in particular for blending, to account for limitations in ignition delays and evaporation properties of the fuel molecules discussed up to now [19].
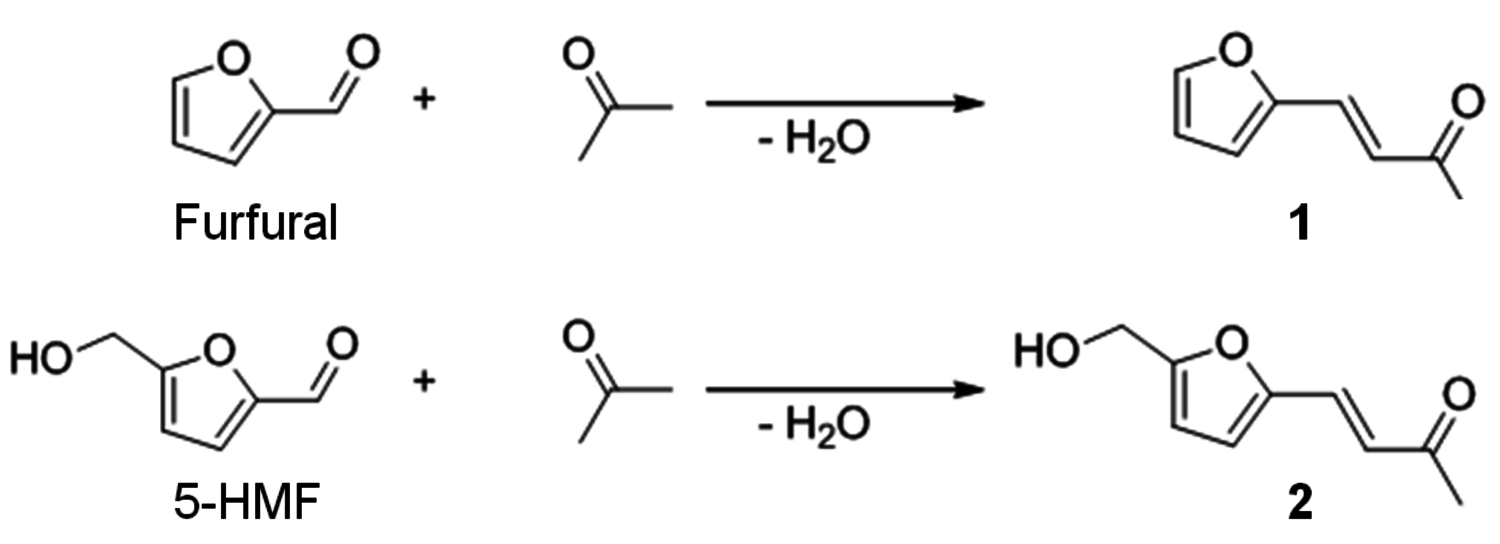
Fig. 12.10: Aldol-condensation of biomass-derived furfuryl derivatives with acetone
Selective hydrogenation of the primary products 1 and 2 gives access to a range of increasingly saturated structures. Deep hydrogenation to the corresponding alkanes has been achieved by heterogeneously catalyzed reactions for both furfuryl derivatives and proposed as a selective route to individual linear alkanes [20]. The intermediate hydrogenation products are also of interest, as they offer various options for further derivatization depending on the remaining functionalities (aromatic, olefinic, C=O, C–OH). Figure 12.11 shows possible hydrogenation products that have not been deoxygenated, highlighting the challenge for the development of selective catalysts and processes to control their formation.
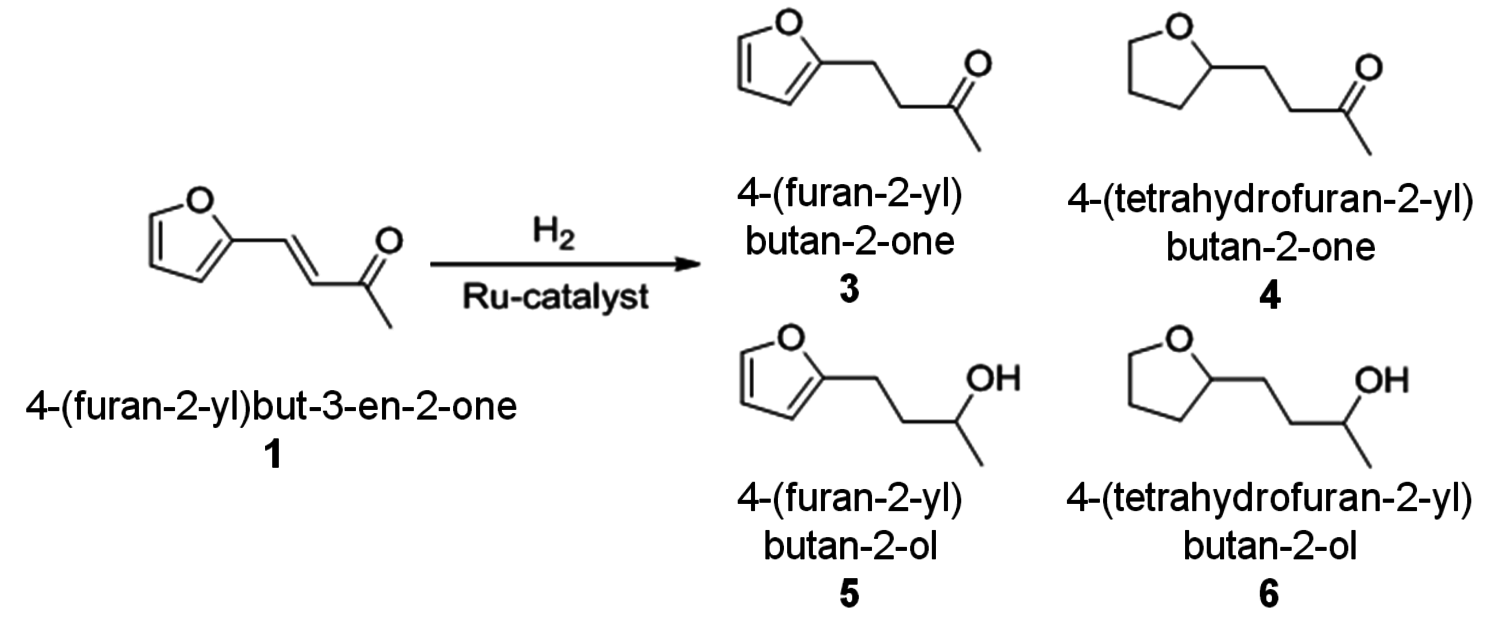
Fig. 12.11: Hydrogenated target structures derived from the aldol-condensation product (1) of furfural with acetone (4-(2-furyl)-3-butene-2-one)
For the selective hydrogenation of the biomass-derived 4-(furan-2-yl)but-3-en-2-one (1) catalysts based Ru-nanoparticles stabilized in ionic liquid matrices (Ru@IL) were investigated [21]. The size of the nanoparticles, as well as their catalytic activity and selectivity can be controlled by variation of the respective IL. The novel catalyst systems were highly active and therefore excellent catalysts for the hydrogenation of C=O and C=C bonds, as well as for the hetero-aromatic ring. Moreover, they show interesting selectivities, which differ from classic homogeneous and heterogeneous Ru-catalysts. The catalytic performance of Ru@[C12MIM][BTA] was compared with the catalytic behavior of classic heterogeneous (ruthenium on alumina) and homogeneous ([RuHCl
 ]) catalysts under identical reaction conditions. The Ru@[C12MIM][BTA] catalyst allowed production of either product 4 or 6 with high selectivity, whereas the homogeneous organometallic complex is very selective for the C=C bond hydrogenation to give 3. At high temperature and hydrogen pressure, the classical heterogeneous catalysts Ru on alumina leads to 6 as the major product. At mild conditions, Ru@[C12MIM][BTA] gave a more selective system that allowed access to the individual products in fair to good yields. Monitoring the course of reaction showed that the hydrogenation of 2 with Ru@[C12MIM][BTA] occurred as a consecutive reaction (Figure 12.12). Initially the C=C double bond of the substrate was hydrogenated, followed by the hydrogenation of the heteroaromatic ring and finally the reduction of the carbonyl group.
]) catalysts under identical reaction conditions. The Ru@[C12MIM][BTA] catalyst allowed production of either product 4 or 6 with high selectivity, whereas the homogeneous organometallic complex is very selective for the C=C bond hydrogenation to give 3. At high temperature and hydrogen pressure, the classical heterogeneous catalysts Ru on alumina leads to 6 as the major product. At mild conditions, Ru@[C12MIM][BTA] gave a more selective system that allowed access to the individual products in fair to good yields. Monitoring the course of reaction showed that the hydrogenation of 2 with Ru@[C12MIM][BTA] occurred as a consecutive reaction (Figure 12.12). Initially the C=C double bond of the substrate was hydrogenated, followed by the hydrogenation of the heteroaromatic ring and finally the reduction of the carbonyl group.
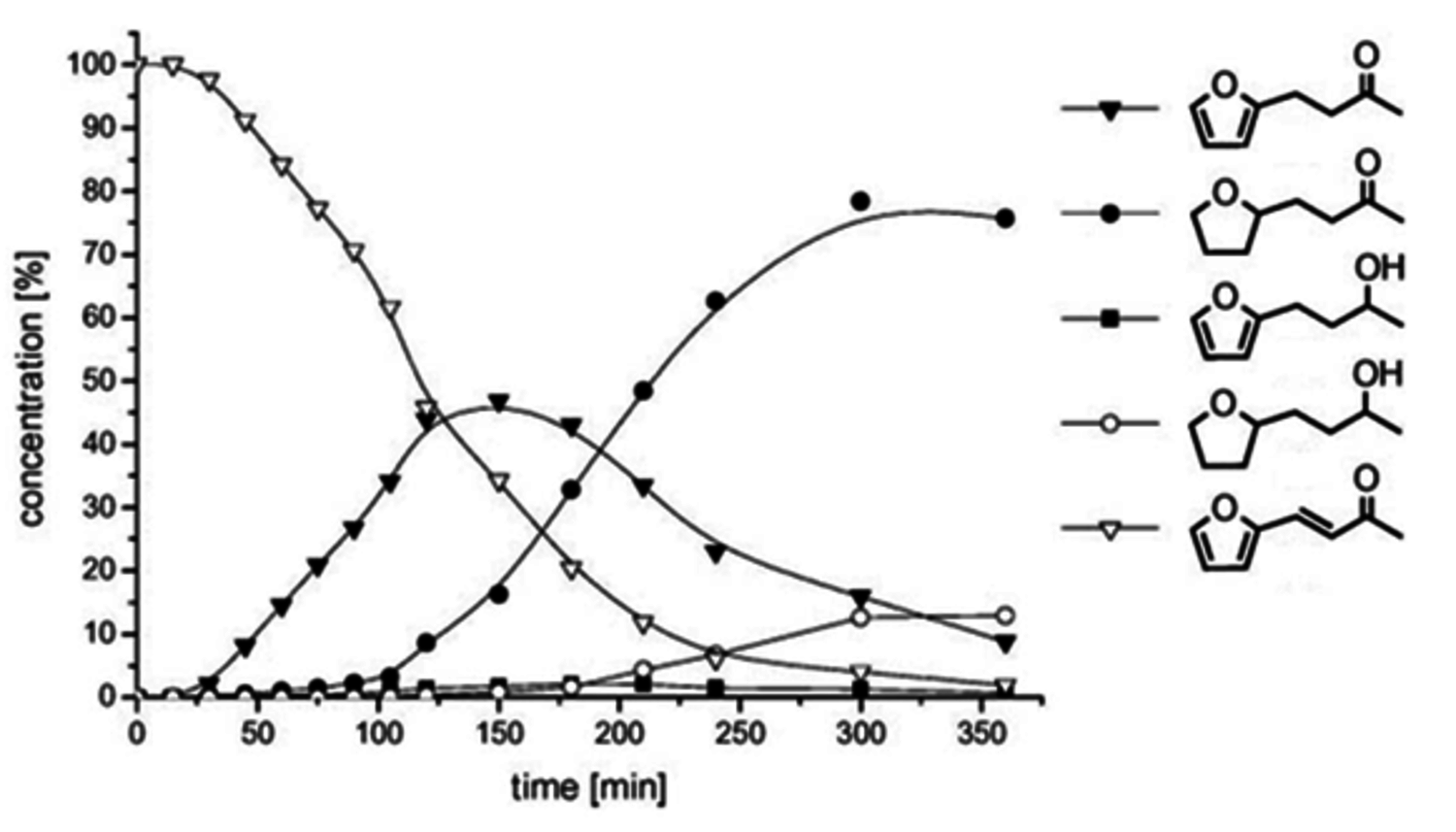
Fig. 12.12: Conversion-time profile for the hydrogenation of 4-(2-furyl)-3-butene-2-one 1 using Ru@[C12MIM][BTA] at T = 60 °C and p(H2) = 60 bar, indicating high selectivity toward product 4. Compound 6 can be obtained in 90% yield under more forcing conditions (T = 120 °C, p(H2) = 120 bar)
The IL-stabilized nanoparticles where found to be readily separated from the products, showing excellent stability and recyclability. The products can be efficiently extracted from the ionic liquid phase by supercritical
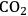 , opening the possibility for continuous-flow processes [13,14]. In repetitive batch-mode, the immobilized ruthenium nanoparticles led to full conversion under standard conditions even after several consecutive cycles. This finding was also supported by TEM measurements, which showed no apparent agglomeration of the nanoparticles. Furthermore, inductively-coupled-plasma mass-spectrometry (ICP-MS) analysis of the isolated products showed a ruthenium contamination below 5 ppm in the product samples, indicating efficient prevention of metal leaching.
, opening the possibility for continuous-flow processes [13,14]. In repetitive batch-mode, the immobilized ruthenium nanoparticles led to full conversion under standard conditions even after several consecutive cycles. This finding was also supported by TEM measurements, which showed no apparent agglomeration of the nanoparticles. Furthermore, inductively-coupled-plasma mass-spectrometry (ICP-MS) analysis of the isolated products showed a ruthenium contamination below 5 ppm in the product samples, indicating efficient prevention of metal leaching.
12.4 From Lignocellulosic Raw Materials to
Carbohydrate Feedstock and Platform Molecules
A preliminary economic and ecolologic analysis of the pathways shown in Figure 12.1 indicates that they provide viable options for sustainable production routes, provided that the platform chemicals are accessible at economic and energetic costs required for typical bio-refinery concepts. The fractionation of the complex and highly functionalized raw material lignocellulose to generate the three main feedstock streams cellulose, hemicellulose and lignin imposes a crucial challenge in this context [22]. As the initial pre-treatment of the raw material represents the entry point into every production route of renewable chemicals and fuels, it is essential to develop highly efficient and selective fractionation processes [23]. This implies the fractionation and separation of hemicellulose (mostly pentose sugars like xylose), cellulose (purely glucose) and the aromatic polymer lignin for subsequent transformation in the production of biofuels, commodities and other high-added-value products. Due to the recalcitrant nature of lignocellulose, current pre-treatment processes are often performed at harsh reaction conditions, leading to concomitant biomass degradation and waste formation. Thus, in order to enable an efficient and sustainable valorization of biomass, an ideal fractionation process should achieve the disentanglement of lignocellulose in a single step, while employing mild reaction conditions and ideally renewable reactants. In a recent approach toward this goal, the catalytic pretreatment of lignocellulose was performed in a biphasic reaction system comprising an aqueous phase containing oxalic acid as catalyst and 2-MTHF as biogenic organic solvent [24]. The conceptual process scheme, combining the selective hydrolysis of hemicellulose with a biphasic separation of lignin is illustrated in Figure 12.13.
In a typical reaction setup, a slurry of beech wood in a biphasic solvent system comprised of dilute aqueous oxalic acid solution (0.1 M) and 2-methyltetra-hydrofuran (2-MTHF) is heated at temperatures ranging from 80–125 °C. Under these conditions the hemicellulose fraction is quantitatively depolymerized to sugar monomers (mainly xylose) via acid catalyzed hydrolysis of the glycosidic bonds. The cellulose fraction is not depolymerized and precipitates as solid residue. Subsequent enzymatic depolymerization of the obtained cellulose pulps with the commercially available enzymatic catalysts, Accellerase-1500 R (Genencor), showed promising activities giving 3.5 g of soluble reducing-end sugars per liter and hour. This indicates that lignin is efficiently removed from the pulp during the process. Indeed, a water insoluble lignin fraction, which is released during the disintegration of the hemicellulose fraction, is extracted into the organic layer and can be isolated by the subsequent removal of the volatile organic 2-MTHF phase.
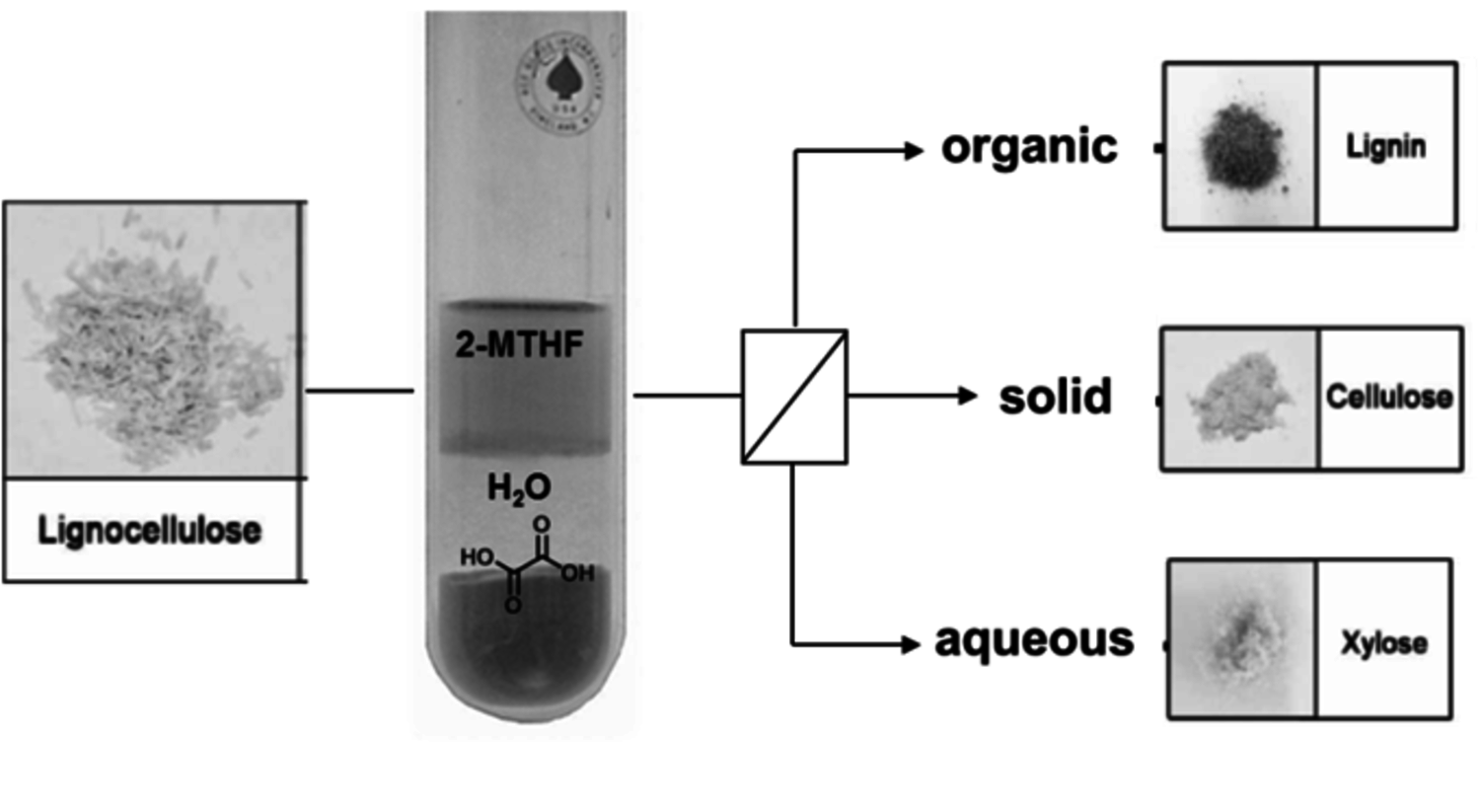
Fig. 12.13: Process scheme of one-step lignocellulose fractionation system
The basic principle of this approach is based on the combination of molecular selectivity and selective extraction of the catalytic system: the applied catalyst oxalic acid is able to depolymerize only the amorphous hemicellulose regions under these conditions, whereas the partly crystalline areas of cellulose-pulp are not cleaved at the applied temperatures. At the same time, the second organic phase enables an efficient in-situ extraction of lignin. With this approach, lignocellulose can be disintegrated into its three major constituents in a single step. With respect to sustainability criteria, this concept benefits from the exclusive use of bio-derived solvents and reagents. Furthermore, recrystallization of the oxalic acid from the aqueous effluent enables straightforward catalyst recycling.
Whatever the most efficient process for the disintegration of biomass ultimately will be, the next important step on the pathway from lignocellulosic raw material to the desired platform chemical relates to the conversion of cellulose (the most abundant renewable feedstock) to glucose. In principle, the catalytic cleavage of the glycosidic bonds in cellobiose or cellulose can be achieved with enzymes or Brønsted acids [25]. However, due to its highly crystalline structure, the essential hurdle is the limited accessibility of cellulose, which is responsible for the low reactivity in such systems. Consequently, reagents able to disrupt the crystalline network before catalytic depolymerization are needed. In this respect, the use of ionic liquids (ILs) in biomass pretreatment enabled the facile disruption of the intrinsic hydrogen bonding between cellulose fibres, and promoted the subsequent dissolution [26].
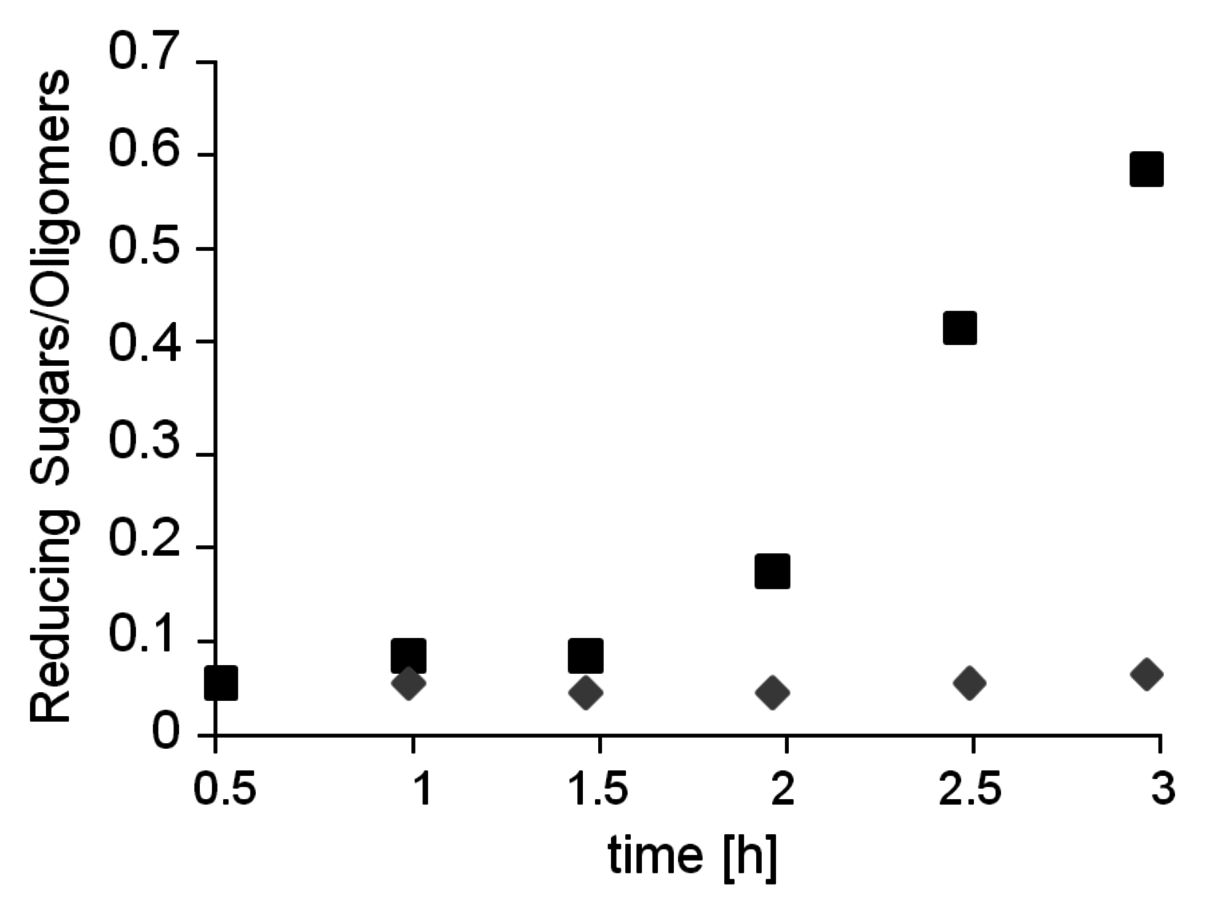
Fig. 12.14: Effect of sodium chloride on hydrolysis of crystalline Avicel®. Diamond: Only oxalic acid. Square: Oxalic acid with 30 wt% NaCl. Reaction conditions: 20 mg Avicel®, 1 mL aqueous NaCl solution (30 wt%), 0.1 M oxalic acid, 105 °C.
Ionic liquids are organic salts with melting points below 100 °C, exhibiting interesting physico-chemical properties that can be tuned widely by molecular design of the cations and anions. Especially acetate and chloride based ILs were found to exhibit unique solvent properties for cellulose and even wooden biomass [27]. Accordingly, ionic liquids greatly enhance the activity in cellulose depolymerization. For example, Schüth et al. reported the combination of the solid acid catalyst Amberlyst 15-Dry and ionic liquid 1-butyl-3-methylimidazolium chloride (BMIMCl), promoting an efficient depolymerization of cellulose to water soluble glucose oligomers under mild conditions [28].
Based on the understanding of the interaction of ILs with the hydrogen bonding network of cellulose, other systems have been developed to mimic the solubility properties with cheaper and more readily recycled materials. Thus, the combination of a simple sodium chloride solution with oxalic acid as catalyst enabled the hydrolysis of highly crystalline Avicel® cellulose at mild reaction conditions (see Figure 12.14, [29]). Even concentrated seawater could be used as processing medium in combination with suitable mild acids. In general, the combination of strong electrolytes that disrupt the hydrogen bonding network of cellulose [30] together with mild acidic media for the selective cleavage of the glycosidic bonds provides an interesting alternative to enzymatic depolymerization processes.
The techniques to produce carbohydrate fractions and feedstocks from lignocellulose are constantly improving and new innovations are emerging, strongly stimulated also by the increasing efforts to implement second generation bio-ethanol on a commercial scale [31]. In addition to these challenges, de-bottleneck- ing is also necessary for the production of the intermediate platform chemicals derived thereof. While the fermentative production of itaconic acid from green biomass is currently in a very preliminary stage [32], levulinic acid and 5-HMF are accessible at least in principle via already established processes, albeit economic constraints are still to be overcome.
Especially levulinic acid is of increasing interest as a primary platform chemical due to its relatively simple production from acid treatment of fructose or glucose (Figure 12.15). The required dehydration reaction is promoted by a number of catalysts, and recent developments include examples ranging from simple Brønsted acids and Lewis acids to heterogeneous catalysts (e.g., zeolites) [33]. The major drawback in this transformation is associated with the thermal instability of the intermediately formed 5-HMF, which tends to polymerize at elevated temperatures. Therefore, the catalyst, reaction media and reaction conditions have to be carefully adapted. In addition, while glucose represents the most abundant hexose, its susceptibility for the dehydration reaction is largely inferior to that of fructose and consequently effective isomerization reactions have been developed in recent years. Especially the combination of chromium dichloride (
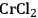 ) and the ionic liquid 1-ethyl-3-methylimidazolium chloride (EMIMCl) showed high conversions for the direct transformation of glucose, presumably via in situ isomerization [34].
) and the ionic liquid 1-ethyl-3-methylimidazolium chloride (EMIMCl) showed high conversions for the direct transformation of glucose, presumably via in situ isomerization [34].
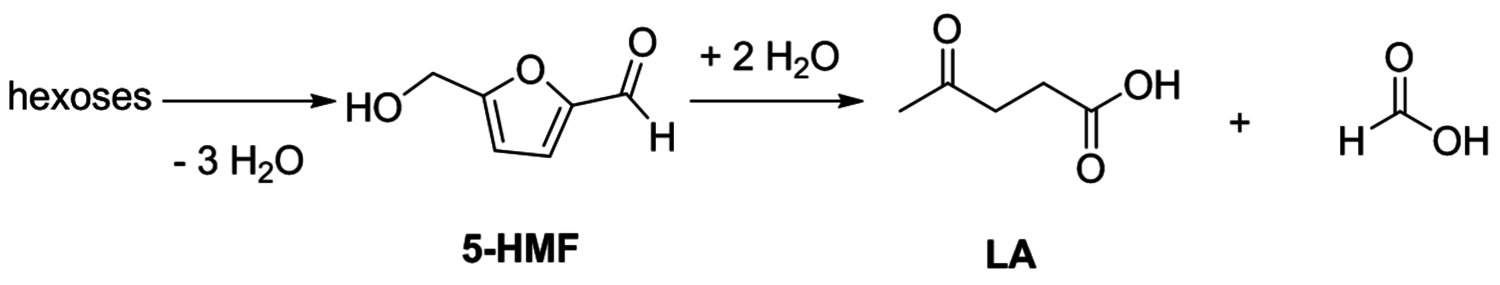
Fig. 12.15: Production of 5-HMF and LA from hexoses
12.5 Conclusion and Outlook
The selective conversion of lignocellulosic biomass provides a possible approach to the sustainable production of fuels and chemical products. Studies of the Cluster of Excellence as well as from other groups around the world demonstrate that the synthetic pathways can be competitive in terms of energy and carbon efficiency to the conventional routes of second generation biofuels. Whereas the latter search mainly for substitutes of existing products, the new approaches aim at improved and tailor-made products. This paradigm shift overcomes the purely limitation-driven (resources or economics) approach to biomass research, opening the field for innovative solutions that may go way beyond a mere feedstock alternative. However, long-term fundamental research is clearly required to reach this ambitious, yet highly attractive goal, which may equally apply to fuels and chemical products.
1Product design: although significant progress has been made in certain areas such as pharmaceuticals, our understanding of the relationship between molecular structures and application properties is still often severely limited for many chemical products. The fuel sector, where properties are largely described via lumped parameters such as octane or cetane numbers, provides a very illustrative example. This becomes even more challenging when several sustainability criteria such as performance, costs, toxicity, and eco-toxicity have to be combined and balanced within a multi-parameter optimization problem.
2Pathway design: The retrosynthetic approach described herein offers a general guideline for the identification and selection of promising pathways. However, the toolbox of classical organic chemistry is currently not widely applicable in these scenarios, as it aims at functionalization, rather than de- and re-functionalization in most cases. Hence, new catalytic methodologies have to be developed, capitalizing on the huge potential of homogeneous, heterogeneous, and bio-catalytic methods. Along the value chain, it is likely that enzymatic and fermentative processes will have to be combined with chemo-catalytic methods in order to create flexible supply networks. As highlighted in the present chapter, the carbohydrate fraction of lignocellulose shows already some promising possibilities in this respect. Lignin, however, the third major constituent of biomass, is fundamentally different and the very heterogeneous structure of the aromatic biopolymer and its linkages make it difficult to envisage a similar strategy based on platform chemicals [35]. Selective bond cleavage reactions have to be developed if defined molecular structures need to be carved out from the complex lignin structures [36].
3Process design: Entering a chemical or energetic supply chain with high molecular weight and highly functional starting materials requires process strategies that are fundamentally different from current refinery concepts. Liquid-phase processes will prevail, and hence separation processes have to be based more on differences in solubility rather than on volatility. Consequently, extraction will gain increasing importance in early stages of the supply chain relative to rectification. The use of advanced fluids such as ionic liquids or supercritical fluids can provide novel approaches for liquid-type processes, albeit the economic constraints are obviously very stringent in this sector. Again, however, several seemingly conflicting aspects have to be balanced, disconnecting for example the classical strategy of economy of scale in refineries from the decentralized logistics of biomass production.
In addition to these specific challenges for the selective conversion approach, the sustainable cultivation and supply with biomass as raw material is a key issue for all scenarios concentrating on renewable resources. Although it is clear that biomass cannot provide enough energy carriers for a full replacement of the entire current fuel consumption, it provides a viable option to address the areas where liquid fuels are inevitable (see Introduction) [37]. Increasing productivity of biomass generation, reducing the land use that might compete with other needs, and any strategy that reduces the consumption of soil and water will all have an enormous impact on the sustainability of a future bioeconomy. Ultimately, however, these necessary improvements can only be exploited if suitable processes are available to convert the raw material into the final products that are required to fulfill the needs of mankind in the energetic and material supply chain.
Acknowledgments
The work from our group described in this conceptual review was funded within the Initiative of Excellence of the German Government as part of the Cluster of Excellence Tailor-Made Fuels from Biomass (TMFB, EXC 236). The fruitful collaboration within the Cluster is gratefully acknowledged, especially with Profs. Wolfgang Marquardt, Stefan Pischinger, Ferdi Schüth, and Regina Palkovits, and Drs. Pablo Dominguez de Maria, Markus Hölscher, Nils Theyssen, Roberto Rinaldi, and Martin Müther.
Bibliography
[1] F.M.A. Geilen, B. Engendahl, B. E., Harwardt B., H. B., A. Harwardt. Selective and Flexible Transformation of Biomass-Derived Platform Chemicals by a Multifunctional Catalytic System Angewandte Chemie International Edition 49: 5510-5514 (2010): 5510-5514.
[2] G.W. Huber, S. Iborra, S. I.. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering Chemical Reviews 106: 4044-4098 (2006): 4044-4098.
[3] A. Voll, W. Marquardt. Reaction Network Flux Analysis: Optimization-Based Evaluation of Reaction Pathways for Biorenewables Processing American Institute of Chemical Engineers Journal (2011):.
[4] L.E. Manzer. Catalytic Synthesis of α-Methylene-γ-Valerolactone: A Bio- mass-Derived Acrylic Monomer Applied Catalysis A: General 272: 249-256 (2004): 249-256.
[5] R.A. Bourne, J.G. Stevens, J. S., Ke J.G.. Maximising Opportunities in Supercritical Chemistry: The Continuous Conversion of Levulinic Acid to γ-Valerolactone in CO Chemical Communications 44: 4632-4634 (2007): 4632-4634.
[6] H. Mehdi, V. Fabos, V. F., Tuba V., T. V., R. Tuba. Integration of Homogeneous and Heterogeneous Catalytic Processes for a Multi-step Conversion of Biomass: From Sucrose to Levulinic Acid, γ-Valerolactone, 1,4-Pentanediol, 2-Methyl-tetrahydrofuran, and Alkanes Topics in Catalysis 48: 49-54 (2008): 49-54.
[7] I.T. Horvath, H. Mehdi, H. M., Fabos H., F. H.. γ-Valerolactone - A Sustainable Liquid for Energy and Carbon-Based Chemicals Green Chemistry 10: 238-242 (2008): 238-242.
[8] L. Deng, J. Li, J. L., Lai J., L. J.. Catalytic Conversion of Biomass-Derived Carbohydrates into γ-Valerolactone without Using an External H Angewandte Chemie International Edition 121: 6651-6654 (2009): 6651-6654.
[9] A. Corma, S. Iborra, S. I.. Chemical Routes for the Transformation of Biomass into Chemicals Chemical Reviews 107: 2411-2502 (2007): 2411-2502.
[10] A.J. Janssen, F.W. Kremer, F. K., Baron F.W., B. F., J.H. Baron. Tailor-Made Fuels from Biomass for Homogeneous Low-Temperature Diesel Combustion Energy Fuels (2011):.
[11] F.M.A. Geilen, B. Engendahl, B. E., Hoelscher B., H. B.. Selective Homogeneous Hydrogenation of Biogenic Carboxylic Acids with [Ru(TriPhos)H](+): A Mechanistic Study Journal of the American Chemical Society 133: 14349-14358 (2011): 14349-14358.
[12] H.-U. Blaser. The Chiral Switch of (S)-Metolachlor: A Personal Account of an Industrial Odyssey in Asymmetric Catalysis Advanced Synthesis & Catalysis 344: 17-31 (2002): 17-31.
[13] U. Hintermair, T. Hoefener, T. H., Pullmann T., P. T.. Continuous Enantioselective Hydrogenation with a Molecular Catalyst in Supported Ionic Liquid Phase under Supercritical CO ChemCatChem 2: 150-154 (2010): 150-154.
[14] U. Hintermair, G. Francio, G. F.. Continuous Flow Organometallic Catalysis: New Wind in Old Sails Chemical Communications 47: 3691-3701 (2011): 3691-3701.
[15] M. Hechinger, A. Voll, A. V.. Towards an Integrated Design of Biofuels and their Production Pathways Computers and Cemical Engineering 34: 1909-1918 (2010): 1909-1918.
[16] F.M.A. Geilen, T. Stein, T. S., vom T., v. T., Engendahl vom, E. v.. Highly Selective Decarbonylation of 5-(Hydroxymethyl)furfural in the Presence of Compressed Carbon Dioxide Angewandte Chemie 123: 6963-6966 (2011): 6963-6966.
[17] G. Tian, R. Daniel, R. D., Li R., L. R., H. Li. Laminar Burning Velocities of 2,5-Dimethylfuran Compared with Ethanol and Gasoline Energy Fuels 24: 3898-3905 (2010): 3898-3905.
[18] Y. Roman-Leshkov, C.J. Barrett, C. B., Liu C.J.. Production of Dimethylfuran for Liquid Fuels from Biomass-Derived Carbohydrates Nature 447: 982-985 (2007): 982-985.
[19] R. Xing, A.V. Subrahmanyam, A. S., Olcay A.V., O. A., H. Olcay, H. O.. Production of Jet and Diesel Fuel Range Alkanes from Waste Hemicellulose-Derived Aqueous Solutions Green Chemistry 12: 1933-1946 (2010): 1933-1946.
[20] E.L. Kunkes, D.A. Simonetti, D. S., West D.A., W. D., R.M. West. Catalytic Conversion of Biomass to Monofunctional Hydrocarbons and Targeted Liquid-Fuel Classes Science 322: 417-421 (2008): 417-421.
[21] J. Julis, M. Hoelscher, M. H.. Selective Hydrogenation of Biomass Derived Substrates using Ionic Liquid-Stabilized Ruthenium Nanoparticles Green Chemistry 12: 1634-1639 (2010): 1634-1639.
[22] B. Kamm, P.R. Gruber, P. G.. Biorefineries—Industrial Processes and Products. Status Quo and Future Directions. Weinheim: Wiley-VCH, 2006
[23] J.-P. Lange. Lignocellulose Conversion: An Introduction to Chemistry, Process and Economics Biofuels, Bioprod. Bioref. 1: 39-48 (2007): 39-48.
[24] T. Stein, Grande vom, G. v., P.M. Grande, P. G., Kayser P.M.. From Biomass to Feedstock: One-Step Fractionation of Lignocellulose Components by Selective Organic-Acid Catalyzed Depolymerization of Hemicellulose in a Biphasic System Green Chemistry 13: 1772-1777 (2011): 1772-1777.
[25] R. Rinaldi, F. Schüth. Acid Hydrolysis of Cellulose as the Entry Point into Biorefinery Schemes ChemSusChem 2(12): 1096-1107 (2009): 1096-1107.
[26] D.A. Fort, R.C. Remsing, R. R., Swatloski R.C., S. R., R.P. Swatloski. Can Ionic Liquids Dissolve Wood? Processing and Analysis of Lignocellulosic Materials with 1-n-Butyl-3-Methlimidazolium Chloride Green Chemistry 9: 63-69 (2007): 63-69.
[27] C. Sievers, M.B. Valenzuela-Olarte, M. VO., Marzialetti M.B., M. M., T. Marzialetti. Ionic Liquid Phase Hydrolysis of Pine Wood Industrial & Engineering Chemistry Research 48: 1277-1286 (2009): 1277-1286.
[28] R. Rinaldi, R. Palkovits, R. P.. Depolymerization of Cellulose by Solid Catalysts in Ionic Liquids Angewandte Chemie 120: 8167-8170 (2008): 8167-8170.
[29] T. Stein, Grande vom, G. v., P. Grande, P. G., Sibilla P., S. P.. Salt-Assisted Organic-Acid-Catalyzed Depolymerization of Cellulose Green Chemistry 12: 1844-1849 (2010): 1844-1849.
[30] A. Pinkert, K.N. Marsh, K. M., Pang K.N.. Ionic Liquids and Their Interaction with Cellulose Chemical Reviews 109: 6712-6728 (2009): 6712-6728.
[31] J. Larsen, M. Ostergaard Petersen, M.O. P., Thirup M. Ostergaard, T. M.O.. The IBUS Process—Lignocellulosic Bioethanol Close to a Commercial Reality Chemical Engineering & Technology 31: 765-772 (2008): 765-772.
[32] J. J. Bozell. Connecting Biomass and Petroleum Processing with a Chemical Bridge Science 329: 522-523 (2010): 522-523.
[33] B.F.M. Kuster. 5-Hydroxymethylfurfural (HMF). A Review Focussing on its Manufacture Starch/Stärke 42: 314-321 (1990): 314-321.
[34] H.B. Zhao, J.E. Holladay, J. H., Brown J.E.. Metal Chlorides in Ionic Liquid Solvents Convert Sugars to 5-Hydroxymethylfurfural Science 316: 1597-1600 (2007): 1597-1600.
[35] J. Zakzeski, P.C.A. Bruijnincx, P. B., Jongerius P.C.A.. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals Chemical Reviews 110: 3552-3599 (2010): 3552-3599.
[36] J.M. Nichols, L.M. Bishop, L. B., Bergman L.M.. Catalytic C–O Bond Cleavage of 2-Aryloxy-1-arylethanols and its Application to the Depolymerization of Lignin Related Polymers Journal of the American Chemical Society 132: 12554-12555 (2010): 12554-12555.
[37] C. Somerville, H. Youngs, H. Y., Taylor H., T. H.. Feedstocks for Lignocellulosic Biofuels Science 329: 790-792 (2010): 790-792.