Chapter structure
- 10.1 Introduction
- 10.2 Thermodynamic Considerations
- 10.3 Formulating Strategies for the Conversion of Sugars to Alkane Fuels: Platform Molecules
- 10.4 The Conversion of Sugars and Polyols to H2 and Alkanes
- 10.5 Conversion of Sugars/Polyols to Liquid Fuels via the Formation of Monofunctional Intermediates
- 10.6 Levulinic Acid and ɣ-Valerolactone Platforms for the Production of Liquid Fuels
- 10.7 Concluding Remarks
- Bibliography
- Footnotes
10.1 Introduction
Biomass is an attractive renewable resource as an alternative to diminishing fossil fuels for the production of hydrogen and liquid transportation fuels. In this chapter, we outline recent processes for the production of hydrogen and fuels from biomass-derived platform molecules, such as sugars, sugar alcohols and derivatives. We start by discussing thermodynamic issues for converting oxygenated hydrocarbons to alkane species. This analysis helps us formulate strategies for the conversion of sugars to alkane fuels. We show that the main strategy to be followed is the deconstruction of lignocellulose through controlled deoxygenation reactions to obtain platform molecules and monofunctional species that retain functionalities for further upgrading reactions, such that molecules with carbon chains longer than the platform molecules (5–6 carbons) can be obtained. These upgrading reactions generally require C–C coupling reactions through reactive functional groups, such as C=C, C=O, and acid groups. We also introduce the concept of catalytic coupling at multiple length scales, to aid in the design of catalysts and/or catalytic systems to achieve cost-competitive biorefining strategies, in which the number of processing steps for reactions and separations is minimized.
10.2 Thermodynamic Considerations
We start this section by discussing some of the thermodynamic issues involved in the conversion of oxygenated hydrocarbons, such as sugars and sugar alcohols derived from biomass to alkane species as presented by Simonetti [1]. Even though hydrogenation of these oxygenated species is very favorable thermodynamically, it is important to remember that the maximum carbon chain length of the alkane product species is limited to the carbon number of the sugar or polyol feed, such as glucose (6 carbon sugar), xylose (5 carbon sugar), sorbitol (6 carbon sugar alcohol) and xylitol (5 carbon sugar alcohol). Therefore, we will address issues involved in the production of alkanes with longer carbon chain lengths than present in the carbohydrate reactant. To illustrate the concepts, we use ethylene glycol as a model reactant, because this molecule is the smallest molecule containing a C–C bond and having a C:O stoichiometry equal to 1:1, representing a sugar alcohol. To study the C–C bond formation step, we investigate the production of butane from ethylene glycol. This thermodynamic analysis is also applicable to the larger C5 and C6 sugars and sugar alcohols derived from lignocellulose to be used as feedstocks for the production of fuels.
The conversion of ethylene glycol to butane can be represented by the following stoichiometric equation:
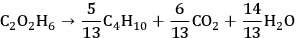
|
10.1 |
This conversion can be described as a combination of two reactions
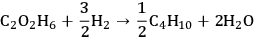
|
10.2 |
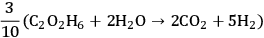
|
10.3 |
where the
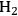 required for the reduction of ethylene glycol (Equation 10.2) is supplied by the reforming of ethylene glycol with water to form
required for the reduction of ethylene glycol (Equation 10.2) is supplied by the reforming of ethylene glycol with water to form
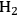 and
and
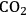 (Equation 10.3). The enthalpy change (at 500 K) for the reduction of ethylene glycol in Equation 10.2 is equal to -37 kcal mol-1, while the enthalpy change for ethylene glycol reforming in Equation 10.3 is equal to 6 kcal mol-1, such that the conversion of ethylene glycol to butane in Equation 10.1 is exothermic by 24 kcal mol-1. Due to the exothermic nature of this reaction, the energy contained in the butane product obtained from 1 mol of ethylene glycol is lower than that of ethylene glycol. The energy content of a compound is expressed by the heat of combustion to form water vapor and
(Equation 10.3). The enthalpy change (at 500 K) for the reduction of ethylene glycol in Equation 10.2 is equal to -37 kcal mol-1, while the enthalpy change for ethylene glycol reforming in Equation 10.3 is equal to 6 kcal mol-1, such that the conversion of ethylene glycol to butane in Equation 10.1 is exothermic by 24 kcal mol-1. Due to the exothermic nature of this reaction, the energy contained in the butane product obtained from 1 mol of ethylene glycol is lower than that of ethylene glycol. The energy content of a compound is expressed by the heat of combustion to form water vapor and
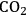 . Figure 10.1 shows the enthalpy changes for the combustion of various compounds that can be derived from ethylene glycol to form water vapor and
. Figure 10.1 shows the enthalpy changes for the combustion of various compounds that can be derived from ethylene glycol to form water vapor and
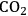 (at a typical reaction temperature of 500 K). The decrease in enthalpy for the conversion of ethylene glycol to butane is sufficiently small that 91% of the energy content of the ethylene glycol reactant is preserved in the alkane product. On the other hand, the mass of butane is approximately 36% of the mass of ethylene glycol, which leads to an increase in energy density. Furthermore, the alkane product is more volatile and less hydrophilic than the oxygenated reactant, which changes the combustion characteristics.
(at a typical reaction temperature of 500 K). The decrease in enthalpy for the conversion of ethylene glycol to butane is sufficiently small that 91% of the energy content of the ethylene glycol reactant is preserved in the alkane product. On the other hand, the mass of butane is approximately 36% of the mass of ethylene glycol, which leads to an increase in energy density. Furthermore, the alkane product is more volatile and less hydrophilic than the oxygenated reactant, which changes the combustion characteristics.

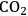 and water vapor have zero energy content. Adapted from [1]
and water vapor have zero energy content. Adapted from [1]Fig. 10.1: Energy content (negative value of enthalpy of combustion at 500 K, kcal mol-1) for various compounds derived from ethylene glycol. The co-products of the combustion,
and water vapor have zero energy content. Adapted from [1]
The conversion of two ethylene glycol molecules to ethanol molecules results in an increase in the energy content of the molecules, from 537 kcal mol-1 to 611 kcal mol-1. This conversion also requires the use of
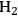 (which has an energy content of 58 kcal mol-1), which must be provided by reforming of ethylene glycol to produce
(which has an energy content of 58 kcal mol-1), which must be provided by reforming of ethylene glycol to produce
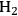 and
and
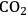 according to Equation 10.6. The stochiometric equation for the conversion of ethylene glycol to ethanol is given by
according to Equation 10.6. The stochiometric equation for the conversion of ethylene glycol to ethanol is given by
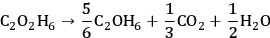
|
10.4 |
which is a combination of the following two reactions:
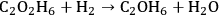
|
10.5 |
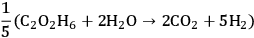
|
10.6 |
Equation 10.5 is exothermic by 21 kcal mol-1 and Equation 10.6 is endothermic by 4 kcal mol-1. As a result, the conversion of ethylene glycol to ethanol according to Equation 10.4 has an enthalpy change of -14 kcal mol-1. With a high equilibrium constant due to the exothermic nature of the reaction, high conversions are achievable. The conversion of ethylene glycol to ethanol demonstrates that the more reduced product has a higher energy content per mol compared to the reactant. Moreover, heat integration can be achieved in a single catalytic reactor by the coupling of the endothermic heat for the reforming reaction to produce
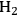 with the exothermic heat of the reduction step. The conversion of ethanol to ethane, similar to the ethylene glycol conversion to ethanol, is a combination of reduction and reforming reactions and results in an increased energy content of ethane compared to ethanol, as can be seen in Figure 10.1.
with the exothermic heat of the reduction step. The conversion of ethanol to ethane, similar to the ethylene glycol conversion to ethanol, is a combination of reduction and reforming reactions and results in an increased energy content of ethane compared to ethanol, as can be seen in Figure 10.1.
As mentioned earlier with respect to the reduction reactions discussed above, the carbon chain length of the alkane product will be the same as that of the reactant. For instance, starting with glucose, the longest carbon chain alkane that can be obtained is hexane. Similar to the case for ethylene glycol conversion, the reduction of glucose to hexane is exothermic, and the reforming of glucose with water to form the required
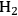 for the reduction reaction is endothermic, such that the overall conversion of glucose to hexane,
for the reduction reaction is endothermic, such that the overall conversion of glucose to hexane,
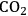 and
and
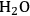 is exothermic (-45 kcal per mole of glucose). If the endothermic energy for the reforming reaction is provided by the exothermic reduction reaction, then the liquid hexane product retains 93% of the energy content of the glucose reactant and only 30% of the original mass of glucose. Even though this conversion to hexane represents a significant increase in the energy density, larger alkanes are required for gasoline, jet, and diesel fuel applications. Therefore, it is now important to focus on reactions employed to increase the length of the alkane chain. At first, we look at the conversion of ethane to butane by the following reaction:
is exothermic (-45 kcal per mole of glucose). If the endothermic energy for the reforming reaction is provided by the exothermic reduction reaction, then the liquid hexane product retains 93% of the energy content of the glucose reactant and only 30% of the original mass of glucose. Even though this conversion to hexane represents a significant increase in the energy density, larger alkanes are required for gasoline, jet, and diesel fuel applications. Therefore, it is now important to focus on reactions employed to increase the length of the alkane chain. At first, we look at the conversion of ethane to butane by the following reaction:
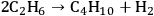
|
10.7 |
As can be seen from Figure 10.1, the conversion of ethane to butane is an endothermic reaction (10 kcal mol-1). Due to the endothermic nature of this reaction, the equilibrium constant would be low, such that high conversions would not be achievable in a single-pass reactor. Another possibility to obtain butane from ethane is oxidative coupling by producing water as a by-product, as demonstrated below:
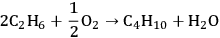
|
10.8 |
This reaction is exothermic with an enthalpy change of -47 kcal mol-1, resulting in a favorable equilibrium constant. However, in practice, it is difficult to achieve high yields of butane because of the high reactivities of alkanes to undergo combustion reactions in the presence of
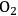 . As demonstrated, the production of longer chain alkane molecules by coupling of light alkane molecules remains a challenge in the field of heterogeneous catalysis.
. As demonstrated, the production of longer chain alkane molecules by coupling of light alkane molecules remains a challenge in the field of heterogeneous catalysis.
By studying Figure 10.1, it can be seen that various coupling reactions involving partially oxygenated reactants are exothermic in nature. Such reactions have favorable equilibrium constants, while not requiring the use of
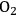 as an oxidizing agent. One example is the coupling between ethane and ethanol to form butane and water (-12 kcal mol-1):
as an oxidizing agent. One example is the coupling between ethane and ethanol to form butane and water (-12 kcal mol-1):
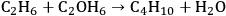
|
10.9 |
The coupling between two ethanol molecules leads to butanol and water in another exothermic reaction (-16 kcal mol-1):
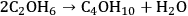
|
10.10 |
This butanol product can then undergo dehydration to butene, followed by hydrogenation to butane, which in the end will lead to an increase in the energy content (from 595 kcal mol-1 to 635 kcal mol-1). The dehydration can be accomplished with high conversion due to the positive entropy change. The conversion of butene to butane then requires the addition of
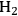 , and in an ideal case, the endothermic production of the required
, and in an ideal case, the endothermic production of the required
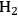 by reforming (accompanied by the production of
by reforming (accompanied by the production of
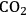 ) would be coupled with the exothermic heat of the olefin hydrogenation step.
) would be coupled with the exothermic heat of the olefin hydrogenation step.
In addition to the coupling of an ethanol molecule with itself or an ethane molecule, other catalytic scenarios can be imagined to achieve C–C coupling to obtain longer carbon chain alkanes. For instance, two molecules of ethanol can undergo dehydrogenation to form acetaldehyde molecules, which can be coupled by aldol condensation. The aldol product can then be dehydrated and hydrogenated to form the alkane product [2]. It is also possible to dehydrate ethanol to obtain ethylene, coupled with ethylene dimerization followed by hydride transfer with ethane over acid catalysts [3].
Comparing the C–C coupling reactions of light alkanes to those of partially oxygenated compounds, we can conclude that partially oxygenated compounds offer new routes for C–C coupling that are unavailable for C–C coupling reactions between alkanes. Thus, the strategy that should be followed to produce alkanes with longer carbon chains than the biomass derived carbohydrate feeds is to utilize the oxygen-containing moieties in the functional reaction intermediates to form C–C bonds, prior to formation of the final alkane product.
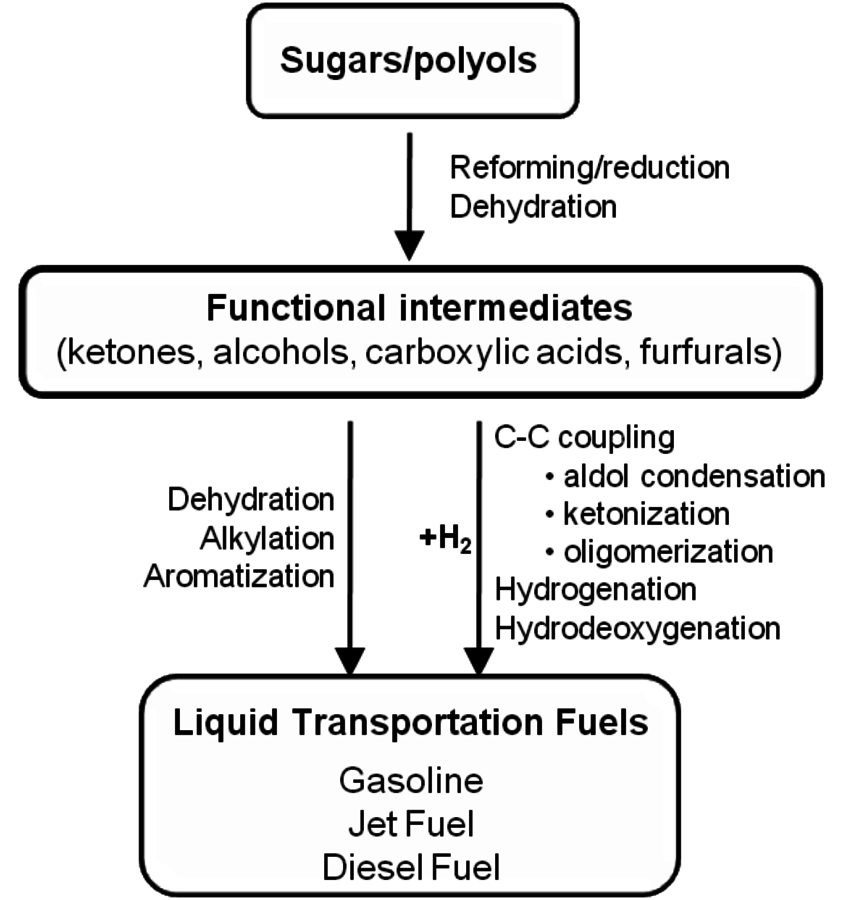
Fig. 10.2: The overall strategy for the production of liquid fuels from sugars and sugar alcohols. Adapted from [1]
These functional intermediates can be obtained from carbohydrates by the combination of reforming and reduction reactions, and/or by selective dehydration processes. These intermediates can then be converted directly to liquid transportation fuels through processes that do not require the addition of hydrogen, such as dehydration, alkylation and aromatization reactions to produce alkanes, olefins, and aromatic compounds. Alternatively, some intermediates might have desired functionalities to undergo C–C coupling reactions, such as aldol-condensation and ketonization processes, followed by hydrogenation and hydrodeoxygenation reactions to produce higher molecular weight alkanes. The
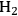 required for these hydrogenation and hydrodeoxygenation steps could be generated by the reforming of the carbohydrate feed. This overall strategy for the production of liquid fuels from carbohydrates is demonstrated in Figure 10.2.
required for these hydrogenation and hydrodeoxygenation steps could be generated by the reforming of the carbohydrate feed. This overall strategy for the production of liquid fuels from carbohydrates is demonstrated in Figure 10.2.
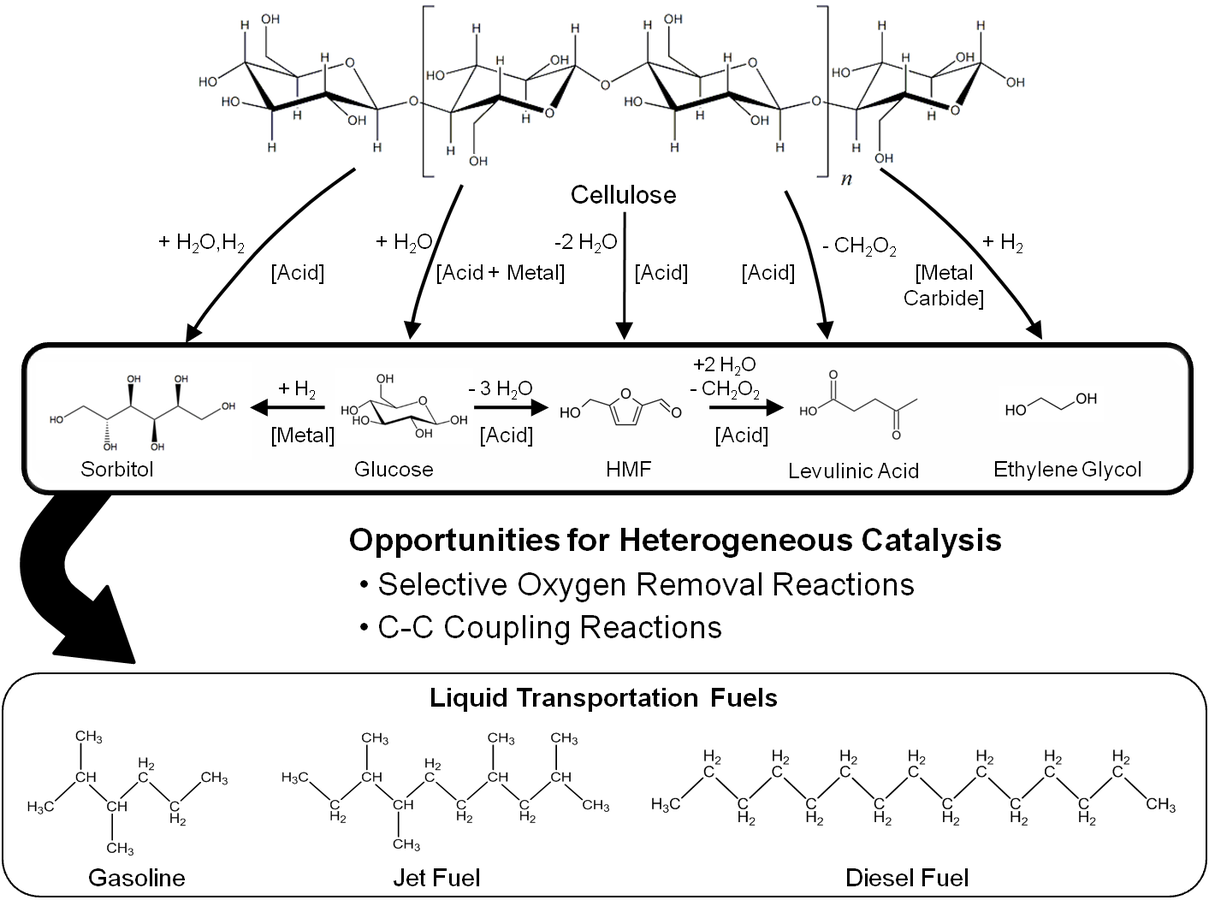
Fig. 10.3: Examples of platform molecules that can be obtained from cellulose and the overall strategy for the production of liquid fuels from biomass through the formation of platform molecules. HMF-5-Hydroxymethylfurfural
10.3 Formulating Strategies for the Conversion of Sugars to Alkane Fuels: Platform Molecules
As discussed in the previous section, a useful strategy for the production of liquid fuels from biomass is to start with controlled oxygen removal reactions to produce functional intermediates, which have sufficient functional moieties to carry out C–C bond forming reactions to obtain long carbon chain alkanes.
Fig. 10.4: Examples of main chemical reaction classes involved in the production of transportation fuels and chemicals from carbohydrate based feedstocks in a bio-refinery. HMF-5-Hydroxymethylfurfural; DHMF-Di(hydroxymethyl)furan. Adapted from [4]
Prior to obtaining these less-reactive intermediates that can be subsequently upgraded to the desired products, biomass (cellulose, hemicellulose and lignin) needs to be converted to molecules that are easier to process but at the same time have sufficient functionalities to allow the production of a wide range of simpler molecules. These functional intermediates can also be referred to as platform molecules, some examples of which include glucose, sorbitol, 5-hydroxymethyl- furfural and levulinic acid. Figure 10.3 demonstrates some platform molecules that can be obtained from cellulose and summarizes the strategy for producing liquid fuels from biomass through the formation of platform molecules. Several reactions can be carried out to obtain these platform molecules and convert them into liquid fuels. In this section, we will outline the main reactions that would be required in biorefining strategies. Figure 10.4 shows some examples of these reactions.
10.3.1 Reaction Classes
1Hydrolysis: Hydrolysis is a major reaction adopted in many biorefining strategies starting from lignocellulosic biomass. It is generally carried out in upstream processing to cleave the glycosidic bonds between the sugar units in cellulose and hemicellulose to obtain simple sugar molecules. Hydrolysis reactions can be carried out using acid or base catalysts at temperatures ranging from 370–570 K, depending on the structure and nature of the polysaccharides. Hydrolysis of cellulose, the most abundant polysaccharide and the major component of lignocellulose, is carried out mostly by using enzymes or dilute mineral acids. High yields of glucose (>90% of theoretical maximum) can be achieved by enzymatic hydrolysis of cellulose after biomass pretreatment [5,6]. Harsher conditions using solutions of mineral acids (
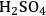 ) at elevated temperatures can be applied to hydrolyze cellulose to obtain degradation products such as 5-hydroxymethylfurfural (HMF) and levulinic acid, which are also platform molecules [7,8]. Hemicelluose can be hydrolyzed at more modest temperatures and dilute acid concentrations, thereby minimizing degradation of the simple sugars obtained, consisting mainly of xylose [8].
) at elevated temperatures can be applied to hydrolyze cellulose to obtain degradation products such as 5-hydroxymethylfurfural (HMF) and levulinic acid, which are also platform molecules [7,8]. Hemicelluose can be hydrolyzed at more modest temperatures and dilute acid concentrations, thereby minimizing degradation of the simple sugars obtained, consisting mainly of xylose [8].
2Dehydration: Dehydration reactions take place in the presence of acid catalysts and result in the formation of C=C bonds following the loss of a water molecule. For instance, sugar molecules can be dehydrated to generate furan compounds such as furfural and HMF, which have functional groups suitable for further upgrading reactions to obtain liquid fuel additives [9]. Another example is the dehydration of alcohols to obtain alkene molecules that can be converted to liquid fuels through oligomerization reactions [10,11].
3Hydrogenation: Hydrogenation reactions are used to saturate C=C and C=O bonds in molecules over metal catalysts, such as Pd, Pt, Ni, or Ru. Some examples in biorefining strategies include hydrogenation of sugars to sugar alcohols and ketones and aldehydes to secondary and primary alcohols, respectively. When hydrogenation is coupled with dehydration over a bifunctional acid/metal catalyst, high molecular weight ketones, aldehydes and alcohols can be converted to alkanes suitable for fuel applications [4,12].
4Hydrogenolysis: Cleavage of C–C and C–O bonds by hydrogen is referred to as hydrogenolysis. This reaction requires the presence of a metal catalyst, such as Pd, Pt, Ni, Ru or Cu and can be carried out under basic conditions [13]. Selective C–O bond cleavage can be used for controlled oxygen removal from highly oxygenated species to obtain monofunctional species such as carboxylic acids and alcohols [14].
5Reforming: Reforming reactions involve the production of CO and hydrogen from carbohydrates by the cleavage of C–C bonds of the carbohydrate backbone. The mixture of CO and
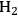 is referred to as synthesis gas and can be used for methanol synthesis as well as for the production of liquid fuels via Fischer-Tropsch synthesis. In the presence of water, water-gas shift reaction can take place to convert CO to
is referred to as synthesis gas and can be used for methanol synthesis as well as for the production of liquid fuels via Fischer-Tropsch synthesis. In the presence of water, water-gas shift reaction can take place to convert CO to
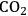 while generating additional
while generating additional
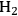 . Steam reforming is utilized to generate hydrogen from alkanes (especially methane) and carbohydrates, whereas aqueous-phase reforming is favorable for hydrogen production starting from aqueous solutions of sugars and sugar alcohols [15]. The thermodynamic and kinetic considerations for hydrogen production by aqueous-phase reforming are discussed in the following sections.
. Steam reforming is utilized to generate hydrogen from alkanes (especially methane) and carbohydrates, whereas aqueous-phase reforming is favorable for hydrogen production starting from aqueous solutions of sugars and sugar alcohols [15]. The thermodynamic and kinetic considerations for hydrogen production by aqueous-phase reforming are discussed in the following sections.
6C–C coupling: As presented earlier, C–C bond formation reactions are crucial for the production of liquid fuels with appropriate molecular weights for gasoline, diesel fuel and jet fuel applications, starting with biomass-derived feedstocks. The most common C–C coupling reactions to be used in a biorefinery are Fischer-Tropsch synthesis, aldol condensation, ketonization and oligomerization. These reactions are shown with representative species in Figure 10.5.
In Fischer-Tropsch synthesis, CO is catalytically hydrogenated to produce mainly alkanes and alkenes. The mixture of CO and
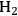 is referred to as synthesis gas and can be produced from reforming reactions. Fischer-Tropsch synthesis is a polymerization reaction; thus, the product stream consists of compounds with a wide molecular weight distribution. This distribution among the product species depends on feed gas composition, pressure, temperature, catalyst and promoters [16,17]. A detailed example will be provided in the following sections for the Fischer–Tropsch synthesis using the synthesis gas generated from the reforming of glycerol.
is referred to as synthesis gas and can be produced from reforming reactions. Fischer-Tropsch synthesis is a polymerization reaction; thus, the product stream consists of compounds with a wide molecular weight distribution. This distribution among the product species depends on feed gas composition, pressure, temperature, catalyst and promoters [16,17]. A detailed example will be provided in the following sections for the Fischer–Tropsch synthesis using the synthesis gas generated from the reforming of glycerol.
Aldol condensation is used to couple two ketone/aldehyde molecules to form a higher molecular weight homologue [18]. This reaction can take place in the presence of either acid or base catalysts and can be used for fine chemical synthesis as well as for the production of fuels. In terms of fuel production, prior to the aldol condensation step, ketones and aldehydes (such as 2-hexanone, acetone, furfural, and HMF) need to be obtained as functional molecules from biomass-derived carbohydrates using controlled C–C and C–O cleavage or dehydration [14,19]. A detailed example will be discussed in the following sections for the conversion of sugars and polyols to monofunctional species such as ketones, alcohols and carboxylic acids over a bimetallic PtRe/C catalyst [14]. The aldol condensation step should be followed by oxygen removal steps to obtain branched or linear, long carbon chain alkanes suitable for gasoline, diesel and jet fuel.
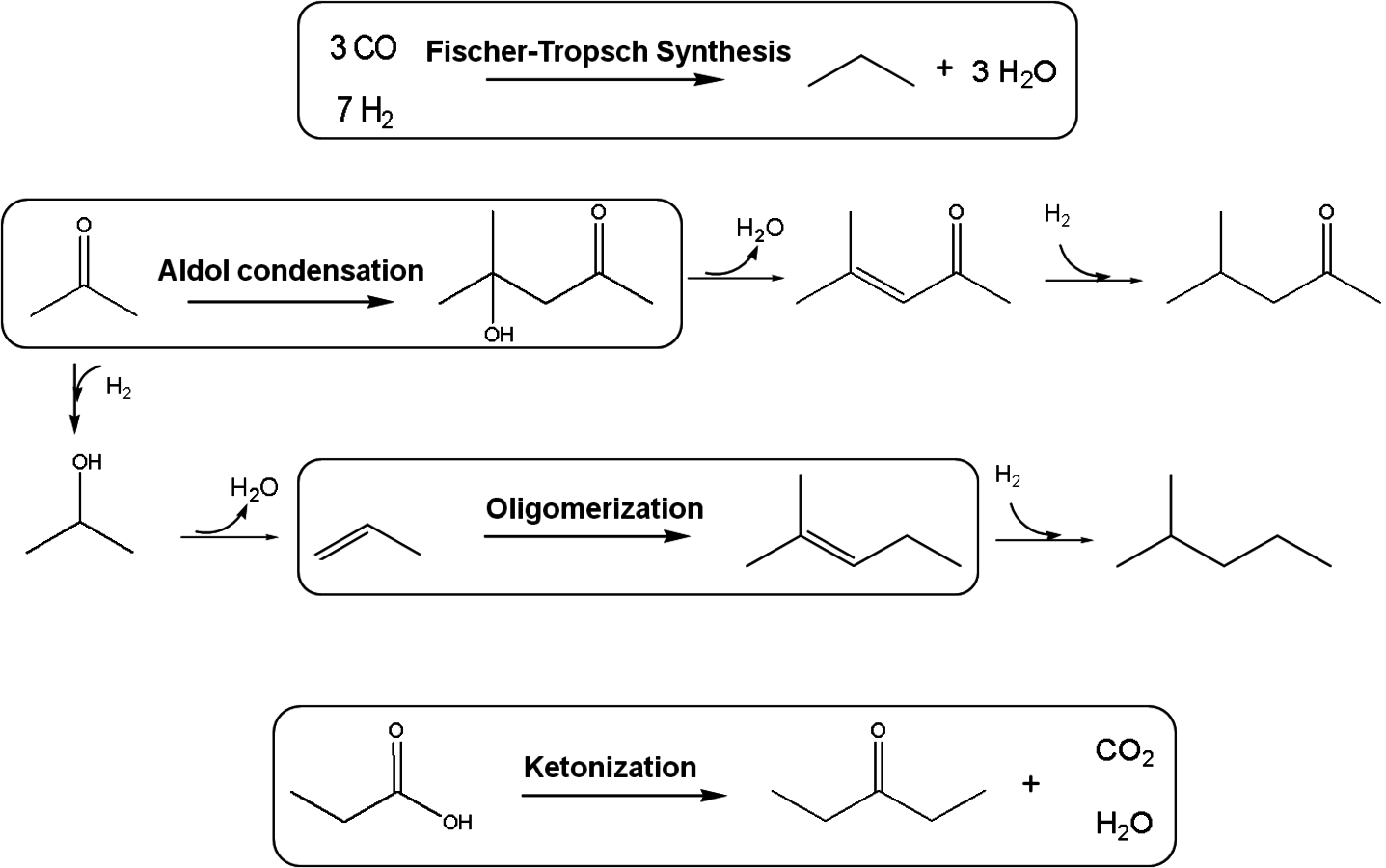
Fig. 10.5: Examples of most common C–C coupling reactions to be used in the production of transportation fuels from carbohydrate based feedstocks in a bio-refinery
In ketonization reactions, two carboxylic acid molecules are combined to form a higher molecular weight ketone molecule, yielding
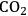 and water mole- cules as the co-products. The product ketone molecules can be converted to alkanes by oxygen removal steps, such as dehydration coupled with hydrogenation. Various studies on the ketonization of carboxylic acids have been presented using several oxides as catalysts such as
and water mole- cules as the co-products. The product ketone molecules can be converted to alkanes by oxygen removal steps, such as dehydration coupled with hydrogenation. Various studies on the ketonization of carboxylic acids have been presented using several oxides as catalysts such as
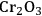 ,
,
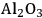 ,
,
 ,
,
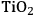 ,
,
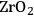 ,
,
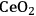 , iron oxide, and manganese oxide, as well as Mg/Al hydrotalcites [20].
, iron oxide, and manganese oxide, as well as Mg/Al hydrotalcites [20].
The oligomerization of light alkenes to produce petrochemicals and fuels is commonly practiced in petroleum-based refineries with high conversion at moderate temperatures (e.g., 470 K). Because it is a well studied and understood reaction, oligomerization is a favorable C–C coupling reaction in biomass processing, making alkenes desirable reaction intermediates [21].
As an example, alkenes can be obtained by dehydration of alcohols in the presence of acid catalysts.
10.3.2 Catalytic Coupling at Multiple Length Scales
The establishment of a biorefinery for the production of fuels and chemicals that is cost competitive with the current petroleum-based refineries requires the development of strategies with a reduced number of processing and separation steps and increased activity and selectivity toward the target products [22]. In order to accomplish these objectives, we consider the possibilities of couplings between catalytic processes at various length scales. Different types of catalytic couplings that will be discussed in further detail are:
1Functional coupling at the active site level,
2active site coupling between different active sites in the same reactor,
3chemical reaction coupling between surface reactions and homogeneous reactions for liquid-phase processes, and
4phase coupling between multiple phases (e.g., aqueous and organic liquids) in liquid-phase processes.
1. Functional Coupling
Functional coupling takes place at the level of the active site and is achieved by chemical bonding interactions that lower the energy of the transition state relative to the reactants and/or products [23]. This lowering in energy, in turn, increases the rate of the overall reaction and the selectivity toward the desired product. Catalytic sites can be created at the subnanometer scale with controlled properties [24]. For instance, active sites can be coupled at the molecular level leading to organic-inorganic hybrid materials (e.g., silica materials functionalized with organic acid and/or base groups) to replace homogeneous acids and bases [24-42]. In these hybrid catalysts two active groups (e.g., an acid and a base) can work cooperatively [34,37-39,43], or a controlled environment around the active center (e.g., changing the steric or electronic environment by incorporating non-polar/polar surface moieties or modifying pore size) can be created [44-52].
One example involves the esterification of fatty acids with alcohols and polyols to form fatty acid esters. The esterification reaction is important for the production of fatty acid esters (which have multiple applications in the food and drug industry), the conversion of vegetable oils and animal fats into biodiesel, and the selective removal of free fatty acids from triglyceride feedstocks [53]. It has recently been demonstrated that the surface hydrophobicity and steric surroundings of the catalytic sites can be tuned to synthesize multifunctional mesoporous silica catalysts for these esterification reactions [50]. For example with glycerol, it is desired to selectively produce monoesters instead of diesters and triesters. Even though conventional homogeneous catalysts, such as p-toluenesulfonic acid, result in high activity, the mono-esters cannot be produced with desired selectivity [50]. On the other hand, high activities coupled with increased selectivity to monoesters can be achieved by using mesoporous silica functionalized with organic sulfonic acid groups. Sulfonic acid groups provide the sufficient acid strength for high rates, while the pore size of the mesoporous silica can be tuned to prevent the formation of bulky diesters and triesters [47-49].
2. Active Site Coupling
Active site coupling can be described by the incorporation of two different types of catalytic sites in the same reactor. This type of catalytic coupling can improve the overall process in different ways. For instance, a rate-controlling reaction step that leads to a reactive intermediate can be coupled with a thermodynamically favorable reaction that converts the reactive intermediate to the final product with increased yield. In such a case, the active sites are generally coupled in a bi- or multifunctional catalyst. One example for this case is the coupling of aldol condensation with hydrogenation using a bifunctional catalyst consisting of base (metal oxide) and metal sites [9,54-57].
This type of approach has been used for the production of methyl isobutyl ketone (MIBK) from acetone. Acetone can undergo aldol condensation over basic catalysts, such as
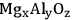 to form diacetone alcohol (DAA) or mesityl oxide (MO) through a consecutive dehydration. MO can later be hydrogenated over a metal catalyst to generate MIBK. However, the production of DAA or MO from acetone is equilibrium limited; and thus, high yields of MIBK cannot be achieved if the hydrogenation takes place in a separate reactor. On the other hand, over a bifunctional catalyst that has both metal and base sites, such as Cu or Pd supported over
to form diacetone alcohol (DAA) or mesityl oxide (MO) through a consecutive dehydration. MO can later be hydrogenated over a metal catalyst to generate MIBK. However, the production of DAA or MO from acetone is equilibrium limited; and thus, high yields of MIBK cannot be achieved if the hydrogenation takes place in a separate reactor. On the other hand, over a bifunctional catalyst that has both metal and base sites, such as Cu or Pd supported over
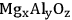 , MO can be hydrogenated to MIBK in the same reactor [55,57]. The conversion of acetone to MIBK is thermodynamically more favorable, and as a result, high yields of MIBK can be obtained in a single stage. The conversion of acetone to MIBK through DAA and MO is shown in Figure 10.6 with corresponding thermodynamic information. Using a bifunctional catalyst not only improves the overall yield of the desired product, but also decreases the number of reaction steps, which in turn decreases the capital and operating costs [9,54,55,57].
, MO can be hydrogenated to MIBK in the same reactor [55,57]. The conversion of acetone to MIBK is thermodynamically more favorable, and as a result, high yields of MIBK can be obtained in a single stage. The conversion of acetone to MIBK through DAA and MO is shown in Figure 10.6 with corresponding thermodynamic information. Using a bifunctional catalyst not only improves the overall yield of the desired product, but also decreases the number of reaction steps, which in turn decreases the capital and operating costs [9,54,55,57].
Another approach for active site coupling is the coupling of two or more reactions in a single reactor, even though individual reactions can be carried out with high yields in separate reactors. Coupling these reactions in a dual-bed or mixed bed system decreases the number of processing steps together with capital and operating costs. The catalytic conversion of glycerol to
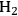 /CO combined with Fischer-Tropsch synthesis for the production of liquid fuels from biomass-derived feedstocks is an example of such a catalytic coupling approach. Because the production of synthesis gas from aqueous glycerol solutions can be accomplished in the same temperature range as Fischer-Tropsch synthesis, the endothermic production of synthesis gas can be coupled with exothermic Fischer-Tropsch synthesis, leading to an energy-integrated process [58,59]. This coupling is described in further detail in the following sections.
/CO combined with Fischer-Tropsch synthesis for the production of liquid fuels from biomass-derived feedstocks is an example of such a catalytic coupling approach. Because the production of synthesis gas from aqueous glycerol solutions can be accomplished in the same temperature range as Fischer-Tropsch synthesis, the endothermic production of synthesis gas can be coupled with exothermic Fischer-Tropsch synthesis, leading to an energy-integrated process [58,59]. This coupling is described in further detail in the following sections.
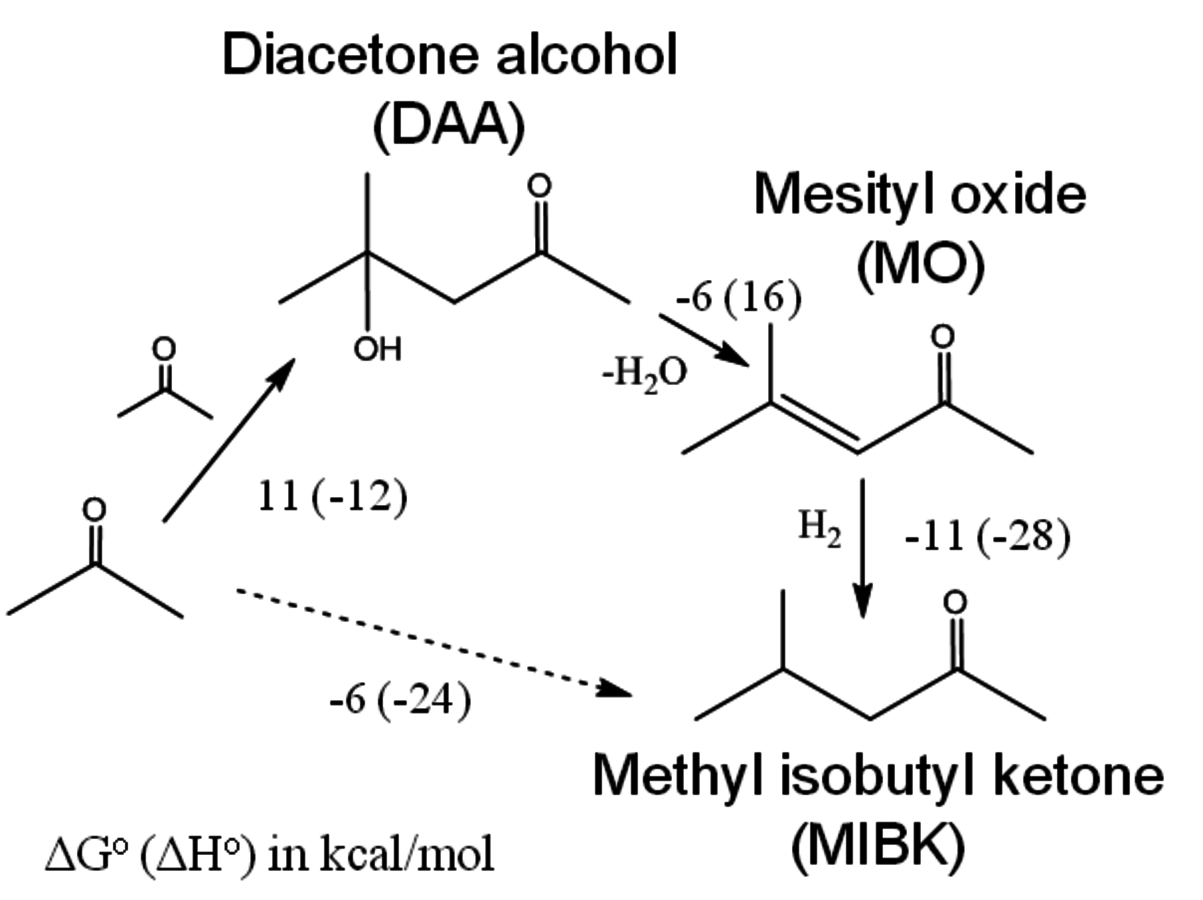
Fig. 10.6: The conversion of acetone to methyl isobutyl ketone (MIBK) through diacetone alcohol (DAA) and mesityl oxide (MO) with corresponding values for ∆G° and ∆H° of reaction (kcal mol-1) of individual steps
3. Chemical Reaction Coupling
As stated earlier, sugar alcohols or polyols, such as glycerol and sorbitol, are common platform molecules, from which fuels and chemicals can be produced. Unlike the production of fuels, the production of fine chemicals requires selective reaction pathways, which can be difficult to achieve due to the high degree of functionality in polyol molecules [60,61]. One example is the hydrogenolysis of glycerol or sorbitol to produce a mixture of glycols and alcohols [60,62-70]. Hydrogenolysis involves multiple reaction steps, such as H-abstraction, C–C bond cleavage and hydrogenation. In addition, the polyol molecules have low volatilities which make them easier to process in the liquid phase than in the gas phase. The presence of these two features opens up the possibility that the conversion of the biomass derived polyols may proceed as a combination of surface reactions as well as homogeneously catalyzed liquid-phase reactions, thereby improving the economics of the process [71-74].
Coupling of surface reactions with homogeneous reactions in the liquid phase is referred to as chemical reaction coupling. In such a case, the reaction in the liquid phase can increase the overall rate of a reaction by lowering the energy barrier for the formation of a key reaction intermediate.
As stated earlier, the aqueous phase hydrogenolysis of polyols leads to a mixture of products resulting from the cleavage of C–O versus C–C bonds [60,64,65]. For instance, the cleavage of C–O bonds in glycerol leads to propanediols, while C–C cleavage generates ethylene glycol and methanol [64,65]. C–O bond cleavages followed by C–C bond breaking can produce mono-alcohols and alkanes [64,65]. According to literature, hydrogenolysis of polyols can go through the following pathways (R1 and R2 represent alkyl groups, hydroxyl groups, alkyl-hydroxyl groups, or H) [75]:
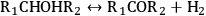
|
10.11 |
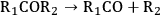
|
10.12 |
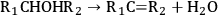
|
10.13 |
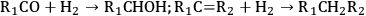
|
10.14 |
Step 1 (Equation 10.11) represents the dehydrogenation of a hydroxyl group to an aldehyde or a ketone, which takes place over metal sites. Step 2 (Equation 10.12) shows the C–C cleavage by retro-aldol reaction, whereas step 3 (Equation 10.13) shows the C–O cleavage by dehydration. Both of these steps are catalyzed by base in the liquid solution. Finally, step 4 (Equation 10.14) represents the hydrogenation of C=O and C=C bonds over a metal catalyst [64,65,68,75]. It is important to note that C–C and C–O bond breaking steps (Equations 10.12 and 10.13) can also be catalyzed by metal sites [64,65].
Recent work from Davis and co-workers on hydrogenolysis of glycerol over both Pt and Ru has provided insight into the roles of surface metal sites and base catalysis in solution [64,65,76]. Even though it is believed that C–C and C–O bond cleavages proceed via base catalyzed retro-aldol reaction and dehydration respectively [75], Davis and coworkers showed that a metal must be present for hydrogenolysis to take place [65]. In these studies, Ru showed a higher overall activity compared to Pt for all conditions tested [65].
The authors also showed that the addition of a base, NaOH in this case, leads to an order of magnitude increase in Ru activity and a 50-fold increase in Pt activity. The authors also reported that the rate of ethylene glycol formation was increased over Pt but stayed unchanged over Ru [65]. In addition, the propylene glycol production rate was increased by an order of magnitude over Ru and by a factor of 50 over Pt [65].
These results demonstrate that C–C bond breaking is metal-catalyzed over Ru and base-catalyzed over Pt, while C–O bond breaking is catalyzed by the base over both metals [65]. Finally, the authors propose that initial dehydrogenation of hydroxyl groups in glycerol is metal-catalyzed and results in the formation of glyceraldehydes. However, the H-abstraction step in dehydrogenation is catalyzed by the base, which in turn enhances the dehydrogenation step [65]. As a result, the mechanism for the hydrogenolysis of glycerol contains chemically coupled steps in which the homogeneous reaction enhances the overall rate in various ways. First of all, the presence of the homogenous catalyst lowers the energy barrier between reactants and transition states by the abstraction of H in the dehydrogenation step. Furthermore, the base-catalyzed retro-aldolization and dehydration reactions generate intermediates for a thermodynamically favorable step (hydrogenation over metal sites).
4. Phase Coupling
In phase coupling, a reaction in one phase generates an intermediate for the reaction in a different phase. Phase coupling can also occur when the reactants and the catalyst exist in different phases. Finally, it is also possible that the second phase is used as an extracting solvent to improve the overall thermodynamics and/or prevent further reaction of the intermediate obtained in the first phase. An example for this approach is the dehydration of sugars to furanic compounds in a biphasic system. High yields can be achieved when monosaccharides, such as glucose, fructose and xylose or polysaccharides, such as sucrose, cellobiose and xylan are dehydrated in the presence of acid catalysts to generate furanic compounds like 5-hydroxymethylfurfural (HMF) and furfural. The dehydration takes place with an acid catalyst, such as HCl or
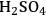 in the aqueous phase, whereas the second phase, a partially miscible organic solvent like butanol, methyl-isobutyl ketone, or dichloromethane, continuously extracts the furanic product to prevent further degradation [2,4,77-79]. It is desirable to utilize low boiling point solvents to eliminate the need of energy intensive separation steps.
in the aqueous phase, whereas the second phase, a partially miscible organic solvent like butanol, methyl-isobutyl ketone, or dichloromethane, continuously extracts the furanic product to prevent further degradation [2,4,77-79]. It is desirable to utilize low boiling point solvents to eliminate the need of energy intensive separation steps.
Detailed studies have been presented in literature for fructose dehydration to generate HMF. The capacity of extracting HMF by the organic phase from the aqueous phase is measured by the ratio of the HMF concentration in the organic phase to that in the aqueous phase after the reaction is completed, and is donated by R. Experimental results suggests a direct relation of HMF selectivity with the value of R [77,78]. Even though the value of R can be increased by using a combination of different organic solvents, it has also been shown that the addition of salt to the aqueous phase can increase R due to the salting-out effect. With the addition of the salt, electrolytes change the intermolecular bonding interactions between liquid components, thereby decreasing the solubility of the two phases in each other. Compared to experiments without salt, a 30 wt% fructose solution saturated with NaCl and 2-butanol as the extracting solvent (with initial ratio of organic and aqueous phase volumes Vorg/Vaq = 1.6) results in an increase in R from 1.6 to 3.3, resulting in an increase in HMF selectivity from 66% to 79% [78]. It is important to note that other than altering the solvent properties, the salt remains inert for the chemical reaction.
10.4 The Conversion of Sugars and Polyols to H2 and Alkanes
In this section, we present a strategy for the production of
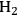 and alkanes from sugar feedstocks. We first look at the thermodynamics of the production of
and alkanes from sugar feedstocks. We first look at the thermodynamics of the production of
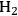 from sugars and polyols in the gas phase as well as in the aqueous phase as demonstrated by Davda et al. [80] previously. Following that, we present kinetic considerations with some examples and discuss how the selectivity toward
from sugars and polyols in the gas phase as well as in the aqueous phase as demonstrated by Davda et al. [80] previously. Following that, we present kinetic considerations with some examples and discuss how the selectivity toward
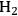 can be shifted toward making alkanes.
can be shifted toward making alkanes.
10.4.1 Thermodynamic Considerations for
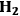 Formation
Formation
The steam reforming reaction of alkanes to generate CO and
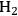 is described in Equation 10.15.
is described in Equation 10.15.
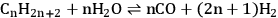
|
10.15 |
Figure 10.7 shows changes in the standard Gibbs free energy (∆G°/RT) for the steam reforming reaction of different alkanes (methane, ethane, propane and hexane), from which it can be seen that the reaction becomes thermodynamically favorable at temperatures higher than 700 K. This temperature is as high as 900 K for the steam reforming of methane. The production of
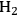 can be increased by the water-gas shift (WGS) reaction, which is shown in Equation 10.16.
can be increased by the water-gas shift (WGS) reaction, which is shown in Equation 10.16.
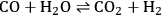
|
10.16 |
The reforming of carbohydrates (C:O ratio being 1:1), such as methanol, ethylene glycol, glycerol and sorbitol, is represented by the equation below (Equation 10.17):
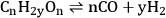
|
10.17 |
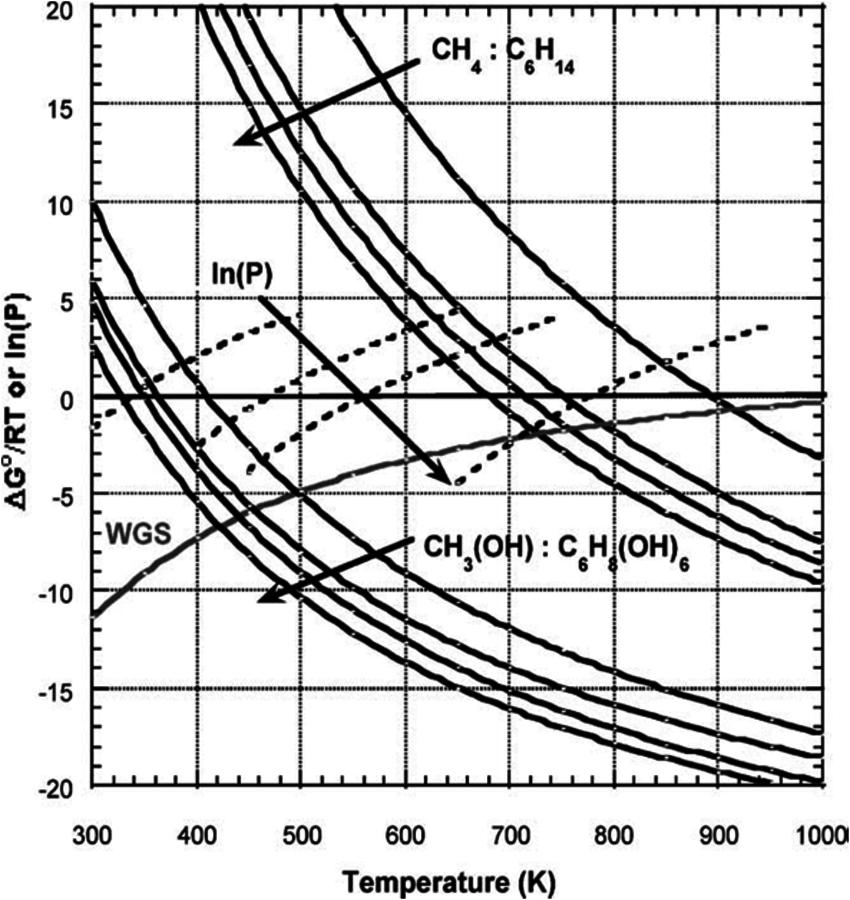
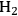 through vapor phase reforming of methane, ethane, propane, hexane, methanol, ethylene glycol, glycerol and sorbitol. Adapted from [80]
through vapor phase reforming of methane, ethane, propane, hexane, methanol, ethylene glycol, glycerol and sorbitol. Adapted from [80]Fig. 10.7: ∆G°/RT values with changing temperature for water-gas shift reaction and production of CO and
through vapor phase reforming of methane, ethane, propane, hexane, methanol, ethylene glycol, glycerol and sorbitol. Adapted from [80]
The value of ∆G°/RT with changing temperature is plotted as well for various carbohydrates in Figure 10.7. Comparison with the values for alkanes shows that the steam reforming of carbohydrates starts to become thermodynamically favorable at considerably lower temperatures. Furthermore, the water-gas shift reaction is also thermodynamically favorable at these lower temperatures. This behavior opens the possibility that hydrogen can be obtained from steam reforming of oxygenated hydrocarbons with a low-temperature route in a single reactor, in which the WGS reaction can also take place. Even though the steam reforming of alkanes generally takes place in the vapor phase, at low temperatures, when WGS is thermodynamically favorable, carbohydrate feeds might have low volatility, which limits the reaction in the vapor phase in return. The vapor phase steam reforming can be carried out for methanol, ethylene glycol and glycerol at temperatures around 500 K. On the other hand, the reforming of heavier carbohydrates such as glucose and sorbitol should be carried out in the liquid phase in order to be coupled with WGS reaction in the same reactor.
Hydrogen production from carbohydrates in the liquid phase is referred to as aqueous phase reforming (APR) and has several advantages over steam reforming. Firstly, the energy requirement is minimized, since neither water nor the carbohydrate feed needs to be vaporized. In addition, as mentioned earlier, the water-gas shift reaction can take place in the same reactor as aqueous reforming and makes it possible to obtain high hydrogen yields with minimal CO levels. Finally, APR takes place at pressures around 15–50 bar where hydrogen purification strategies such as pressure-swing adsorption are effective.
10.4.2 Kinetic Considerations
Even though the thermodynamics of APR reactions seem to be favorable, it is also important to discuss kinetic considerations, such as selectivity issues for this reaction. Firstly, it is known in literature that at low temperatures the formation of alkanes is favorable from
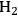 , CO and
, CO and
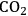 mixtures through methanation and Fischer-Tropsch synthesis reactions [81]. In addition, C–O bond breaking, which leads to alkanes by consecutive hydrogenation reactions, is a competitive pathway to the desired C–C bond cleavage reactions. In short, in order to reach high yields toward
mixtures through methanation and Fischer-Tropsch synthesis reactions [81]. In addition, C–O bond breaking, which leads to alkanes by consecutive hydrogenation reactions, is a competitive pathway to the desired C–C bond cleavage reactions. In short, in order to reach high yields toward
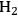 , an efficient catalyst is required that promotes reforming reactions (C–C scission followed by water-gas shift) and inhibits alkane-formation reactions (C–O scission followed by hydrogenation) as well as methanation and Fischer-Tropsch synthesis reactions. A high activity for the water-gas shift reaction is also important in terms of removing CO from the metal surface at low reforming temperatures. Parallel and series reaction pathways that affect the selectivity toward the production of
, an efficient catalyst is required that promotes reforming reactions (C–C scission followed by water-gas shift) and inhibits alkane-formation reactions (C–O scission followed by hydrogenation) as well as methanation and Fischer-Tropsch synthesis reactions. A high activity for the water-gas shift reaction is also important in terms of removing CO from the metal surface at low reforming temperatures. Parallel and series reaction pathways that affect the selectivity toward the production of
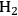 from oxygenated carbohydrate feeds are shown in Figure 10.8.
from oxygenated carbohydrate feeds are shown in Figure 10.8.
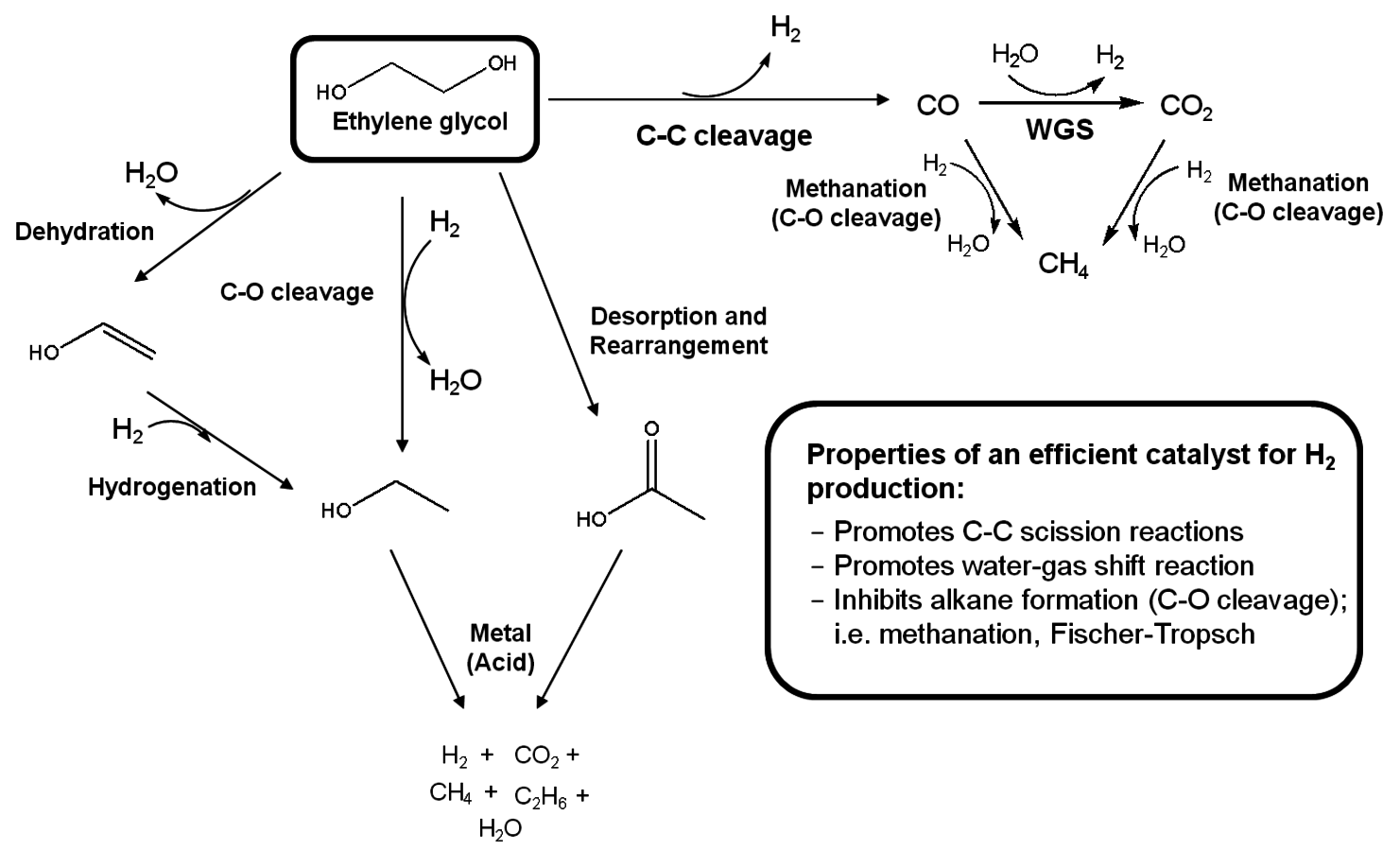
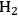 and alkanes over supported metal catalysts. Adapted from [15]
and alkanes over supported metal catalysts. Adapted from [15]Fig. 10.8: Parallel and series reaction pathways involved in the catalytic conversion of biomass-derived oxygenated compounds to
and alkanes over supported metal catalysts. Adapted from [15]
Previous studies on the C–C bond cleavage activity over different metals show that Ru, Ni, Ir and Rh demonstrate high activity followed by Pt [82]. In terms of water-gas shift activity, over an alumina support, Cu exhibits the highest activity followed by Pt, Ru and Ni [83]. Finally, in terms of methanation activity, it has been reported in literature that Ru, Ni and Rh exhibit the highest rates of methanation, whereas Pt, Ir and Pd show lower catalytic activities. When all this kinetic information is combined, it can be concluded that by being active for C–C bond cleavage and water-gas shift reaction as opposed to methanation (C–O cleavage), Pt and Pd should be suitable for the selective production of
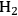 by aqueous-phase reforming of oxygenated carbohydrates [84].
by aqueous-phase reforming of oxygenated carbohydrates [84].
As predicted above, Pt-black and Pt supported on various supports like
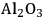 ,
,
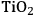 , and
, and
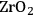 showed high activity and selectivity toward
showed high activity and selectivity toward
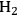 for the APR reaction of methanol and ethylene glycol [80]. Pd supported catalysts have shown similar selectivity, although with a lower activity compared to Pt-based materials. It has also been reported that Rh, Ru, and Ni favor the production of alkanes from polyols over hydrogen [81], because C–O bond cleavage is favored over the C–C bond cleavage over these metals. Experimental results for the APR of glucose, sorbitol, glycerol, ethylene glycol, and methanol over a Pt/
for the APR reaction of methanol and ethylene glycol [80]. Pd supported catalysts have shown similar selectivity, although with a lower activity compared to Pt-based materials. It has also been reported that Rh, Ru, and Ni favor the production of alkanes from polyols over hydrogen [81], because C–O bond cleavage is favored over the C–C bond cleavage over these metals. Experimental results for the APR of glucose, sorbitol, glycerol, ethylene glycol, and methanol over a Pt/
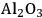 catalyst at 498 and 538 K show that the selectivity for
catalyst at 498 and 538 K show that the selectivity for
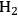 production improves in the order glucose < sorbitol < glycerol < ethylene glycol < methanol.
production improves in the order glucose < sorbitol < glycerol < ethylene glycol < methanol.
Acidity, introduced by the use of solid acid supports (i.e.,
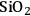 /
/
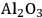 ), increases dehydration rates, which in turn increases the selectivity toward alkanes at the expense of hydrogen [15].
), increases dehydration rates, which in turn increases the selectivity toward alkanes at the expense of hydrogen [15].
In order to describe the kinetics of the reforming of oxygenates in terms of competitive C–C and C–O bond breaking pathways, we provide two example studies (i.e., ethanol and ethylene glycol reforming), in which density functional theory (DFT) calculations have been used together with experimental data.
Example: The Reforming of Ethanol
Ethanol has been chosen for study because it is the simplest oxygenated hydrocarbon, possessing both C–C and C–O bonds. Alcalá et al. [85] carried out DFT calculations to investigate the nature of the surface intermediates and transition states formed from ethanol reforming as a result of C–C and/or C–O bond cleavage pathways over Pt(111) surfaces. The main aim of this investigation was to understand which pathways were most favorable for the reforming of oxygenated hydrocarbons over Pt surfaces.
The DFT studies were initiated by determining the most stable surface intermediates that can be formed from ethanol by removal of hydrogen atoms (no breaking of C–C or C–O bonds) as a first step. Out of many possible species with stoichiometry of
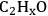 , the most stable species were determined as ethanol, 1-hydroxyethyl (
, the most stable species were determined as ethanol, 1-hydroxyethyl (
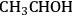 ), 1-hydroxyethylidene (
), 1-hydroxyethylidene (
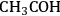 ), acetyl (
), acetyl (
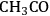 ), ketene (
), ketene (
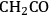 ), ketenyl (CHCO), and CCO species. Following this analysis, surface energies of all the possible products of C–C and C–O bond cleavages (i.e., stabilities of adsorbed O, OH,
), ketenyl (CHCO), and CCO species. Following this analysis, surface energies of all the possible products of C–C and C–O bond cleavages (i.e., stabilities of adsorbed O, OH,
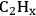 , and
, and
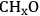 species) were determined. Finally, with the results from DFT calculations and through Brønsted-Evans-Polanyi correlations, the energies of the transition states for C–C and C–O cleavage reactions were determined [85,86]. These correlations relate the energy of the transition state to the energy of the products (final state) with each surface reaction being defined in the exothermic direction. The energies of the transition states (TS) and final states (FS) are relative to the energy of the corresponding initial state in the gas phase. It was determined that among the dehydrogenation products of ethanol on the surface, 1-hydroxyethylidene (
species) were determined. Finally, with the results from DFT calculations and through Brønsted-Evans-Polanyi correlations, the energies of the transition states for C–C and C–O cleavage reactions were determined [85,86]. These correlations relate the energy of the transition state to the energy of the products (final state) with each surface reaction being defined in the exothermic direction. The energies of the transition states (TS) and final states (FS) are relative to the energy of the corresponding initial state in the gas phase. It was determined that among the dehydrogenation products of ethanol on the surface, 1-hydroxyethylidene (
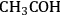 ) species had the lowest energy transition state for C–O bond cleavage, and the ketenyl (CHCO) species had the lowest-energy transition state for C–C bond cleavage.
) species had the lowest energy transition state for C–O bond cleavage, and the ketenyl (CHCO) species had the lowest-energy transition state for C–C bond cleavage.
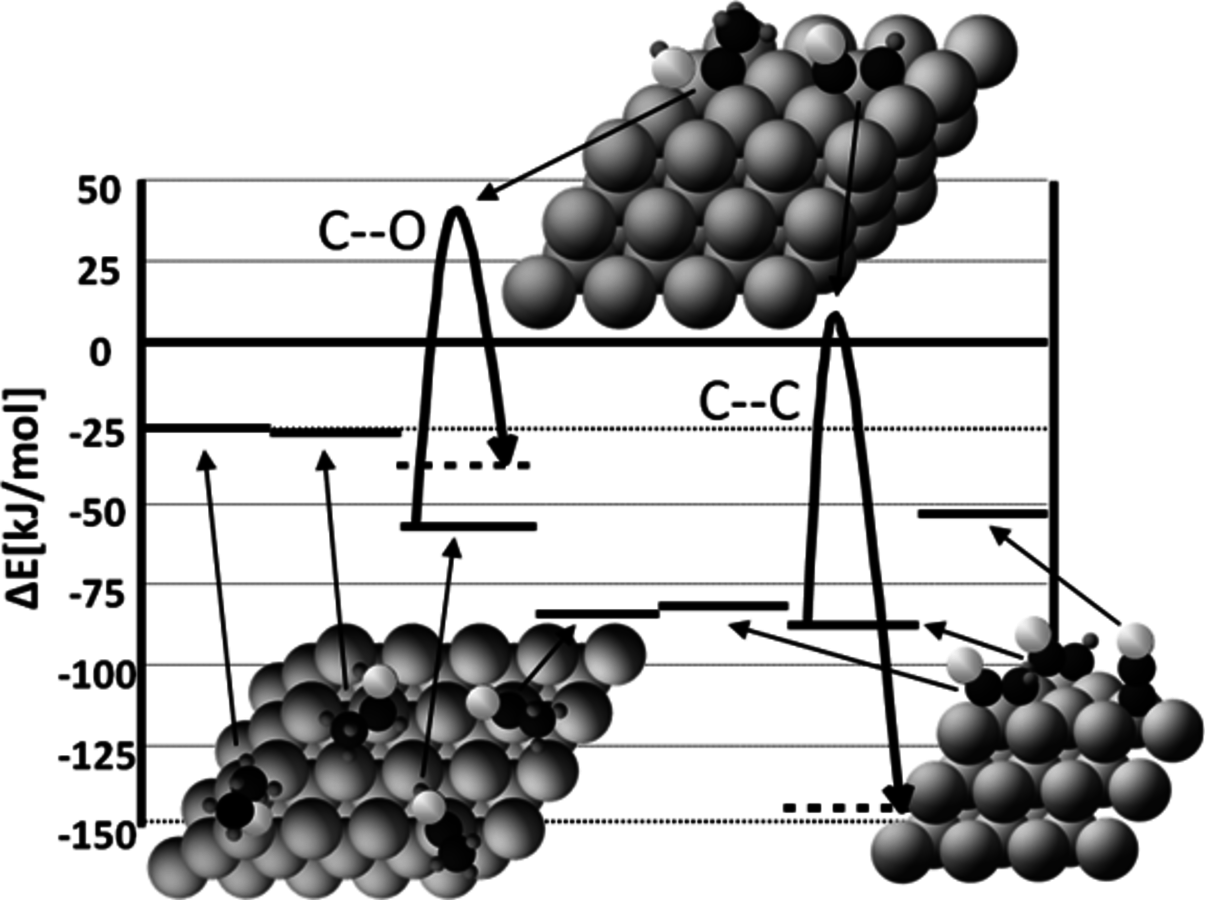
Fig. 10.9: Simplified potential energy diagram of the stabilities and reactivities of dehydrogenated species derived from ethanol on Pt(111). The reference state is gas-phase ethanol and clean slab. Only the most stable species and transition states for C–O and C–C bond cleavage are shown. The black, orange (or light gray) and small blue (or dark grey) balls represent carbon, oxygen and hydrogen atoms, respectively. Adapted from [85]
Figure 10.9 presents a simplified potential energy diagram of the stabilities and reactivities of dehydrogenated species derived from ethanol on Pt(111). Only the most stable species and transition states for C–O and C–C bond cleavage are shown in this schematic potential energy diagram. It can be seen in this figure that C–O bond cleavage occurs on more highly hydrogenated species compared to C–C bond cleavage. In addition, it is shown that the energy of the lowest transition state of C–C bond breaking with respect to ethanol is 4 kJ/mol, whereas it is 42 kJ/mol for C–O bond cleavage. This difference indicates that C–C cleavage should occur at a faster rate compared to C–O cleavage for species derived from ethanol.
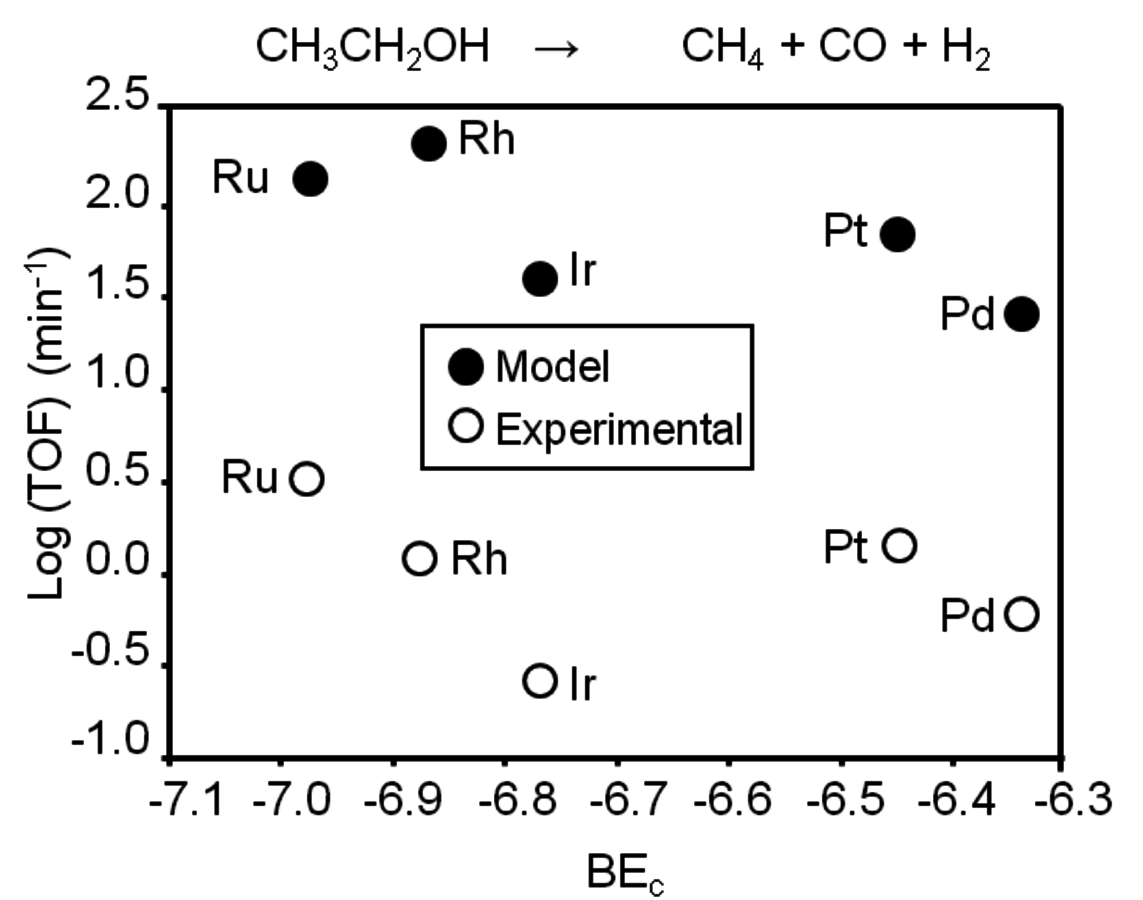
Fig. 10.10: Experimental rates in terms of turnover frequency (TOF) in comparison with the predictions from DFT calculations for C–C bond cleavage of ethanol over Pt, Pd, Ir, Rh and Ru. Open and solid circles show the experimental data and model calculations, respectively. Adapted from [86]
Ferrin et al. [85,86] studied the decomposition of ethanol over various supported metal catalysts, such as Cu, Pt, Pd, Ir, Rh and Ru. The experimental results show that for all the metals except for Cu, the rate of methane and CO formation is 1 to 2 orders of magnitude higher than the rate of the formation of ethane. This behavior indicates that the rate of C–C bond cleavage is considerably higher than that of C–O cleavage, which supports the conclusions reached from DFT studies presented above. Cu does not show activity of C–C or C–O bond cleavage due to low stability of surface intermediates and transition states on the Cu surface. Figure 10.10 presents the experimental rates in terms of turnover frequency (TOF) in comparison with the predictions from DFT calculations for C–C bond cleavage over Pt, Pd, Ir, Rh and Ru. It can be seen from Figure 10.10 that for each metal, the value of the TOF predicted by the model is consistently higher than the value measured experimentally. This difference is acceptable, given the uncertainty in DFT and the correlations used. However, it is evident that the model captures the trends observed in different metals experimentally. It is stated by the authors that the selectivity toward C–C cleavage is due to the weaker bonding of O compared to C on these metal surfaces. With any C–O cleavage step, one of the resulting species will be bound to the surface through the oxygen atom. If binding of this species to the surface is weak, then the final state of C–O cleavage step will be less stable compared to that of C–C cleavage step. This conclusion suggests that in order to be able to cleave the C–O bond selectively, a metal catalyst with stronger binding of O is required.
Example: The Reforming of Ethylene Glycol
Next we look at the kinetics of the reforming of ethylene glycol. Ethylene glycol (EG) is an important model compound to study, because with a C–C bond and having a C:O stoichiometry equal to 1:1, it represents a sugar alcohol. We focus on work carried out by Kandoi et al. [87], in which experimental data (Pt supported on alumina) as well as results of DFT calculations (Pt(111)) are reported for the reforming of EG in vapor as well as in the aqueous phase. Using the experimental and DFT results, microkinetic models were developed for the reaction in both phases, providing insight into the similarities and differences in the reforming chemistries taking place in both phases.
Similar to the analysis of ethanol reforming, DFT calculations were carried out to find the surface energies of all intermediates that can be derived from the dehydrogenation of EG on Pt(111). The most stable
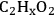 (x being from 0 to 6) species derived from the subsequent dehydrogenation of EG (with respect to EG in the gas phase and clean slab) are
(x being from 0 to 6) species derived from the subsequent dehydrogenation of EG (with respect to EG in the gas phase and clean slab) are
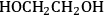 ,
,
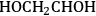 ,
,
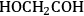 or HOCHCHOH, HOCHCOH, HOCCOH, HOCCO, and OCCO. With the use of Brønsted-Evans-Polanyi type correlations developed for C–H/O–H and C–C bond scissions in other oxygenated compounds on Pt(111) together with DFT calculations, activation barriers for C–C and C–O bond cleavage reactions were calculated over Pt(111). Figure 10.11 shows the Gibbs free energy changes (at 483 K) for these reaction steps (relative to EG in the gas phase and the clean slab). The most stable species resulting from dehydrogenation of EG and the most stable transition states for C–H/O–H and C–C bond scission are included in this figure. The DFT calculations show that at an intermediate value of x (around
or HOCHCHOH, HOCHCOH, HOCCOH, HOCCO, and OCCO. With the use of Brønsted-Evans-Polanyi type correlations developed for C–H/O–H and C–C bond scissions in other oxygenated compounds on Pt(111) together with DFT calculations, activation barriers for C–C and C–O bond cleavage reactions were calculated over Pt(111). Figure 10.11 shows the Gibbs free energy changes (at 483 K) for these reaction steps (relative to EG in the gas phase and the clean slab). The most stable species resulting from dehydrogenation of EG and the most stable transition states for C–H/O–H and C–C bond scission are included in this figure. The DFT calculations show that at an intermediate value of x (around
 ), the activation energy barriers to break the C–C bond become similar to those for C–H/O–H cleavage. This comparison suggests that after this point, C–C cleavage becomes a kinetically competitive pathway.
), the activation energy barriers to break the C–C bond become similar to those for C–H/O–H cleavage. This comparison suggests that after this point, C–C cleavage becomes a kinetically competitive pathway.
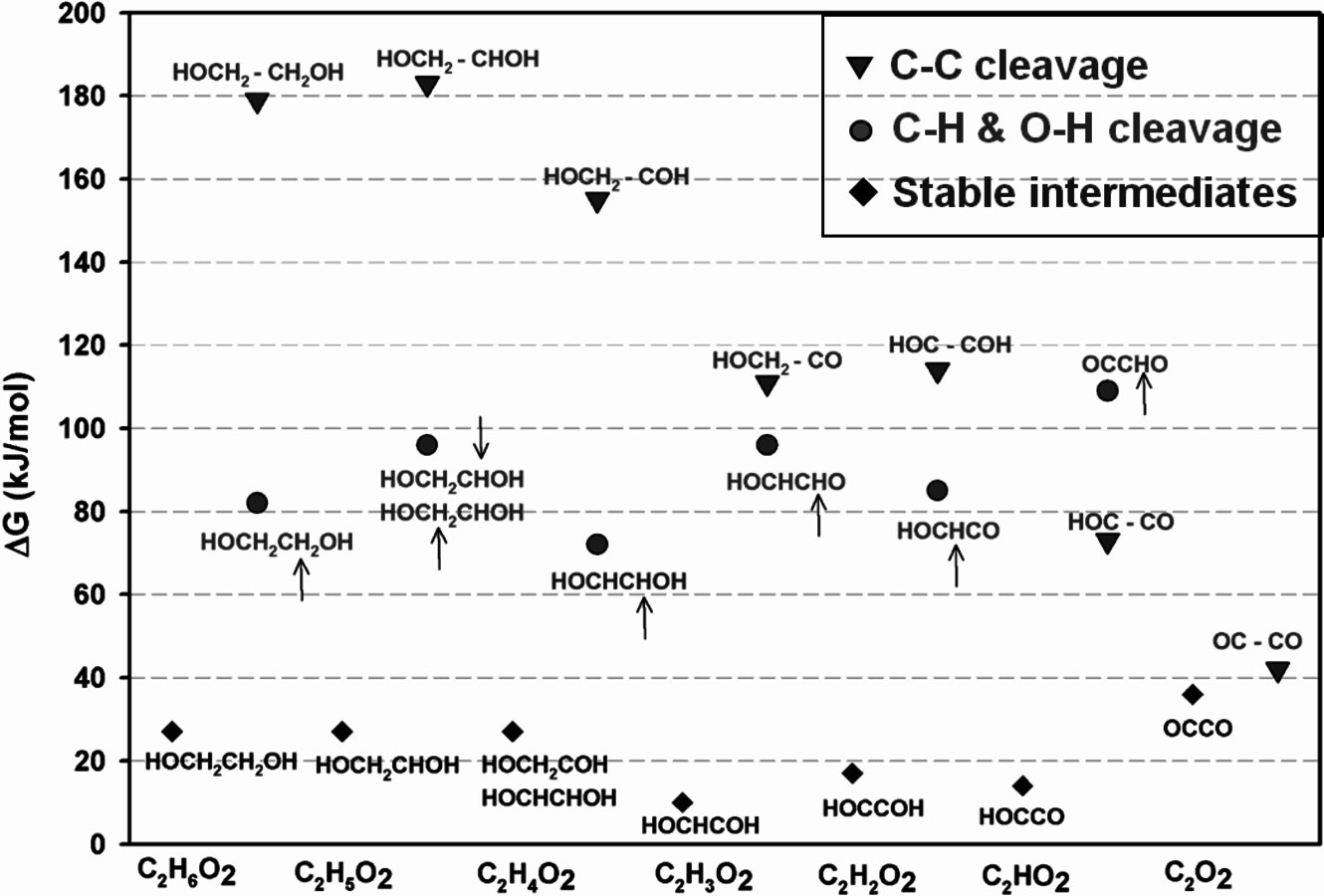
Fig. 10.11: The Gibbs free energy changes (at 483 K) for C–H/O–H and C–C bond scission steps (relative to ethylene glycol in the gas phase and the clean slab) starting from ethylene glycol. The most stable species resulting from dehydrogenation of ethylene glycol (diamonds) and the most stable transition states for C–H/O–H (circles) and C–C bond (inverted triangles) scission are included. The location of C–H cleavage is shown by a small arrow above or below the corresponding hydrogen atom. Adapted from [87]
A microkinetic model, based on the results from the DFT calculations, was developed to understand the vapor and aqueous phase reforming kinetics of EG on Pt. The model was simplified by combining various elementary steps, which resulted in a 7-step reaction mechanism. The simplified mechanism is shown below (Equations 10.18–10.24). Note that, A* and B** denote monodentate and bidentate adsorbed species, respectively.
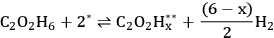
|
10.18 |
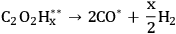
|
10.19 |
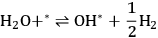
|
10.20 |
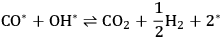
|
10.21 |
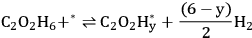
|
10.22 |
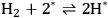
|
10.23 |
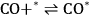
|
10.24 |
Step 1 (Equation 10.18) is the dehydrogenation of EG to generate a surface intermediate
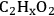 **. This surface species then decomposes into CO and
**. This surface species then decomposes into CO and
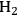 in Step 2 (Equation 10.19). Steps 3 and 4 (Equations 10.20 and 10.21) represent the WGS reaction steps, where CO reacts with
in Step 2 (Equation 10.19). Steps 3 and 4 (Equations 10.20 and 10.21) represent the WGS reaction steps, where CO reacts with
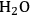 (dissociatively adsorbed) to generate
(dissociatively adsorbed) to generate
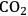 and
and
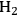 . EG forms a spectator species (
. EG forms a spectator species (
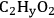 *) in step 5 (Equation 10.22). Finally, Steps 6 and 7 (Equations 10.23 and 10.24) are the adsorption-desorption of
*) in step 5 (Equation 10.22). Finally, Steps 6 and 7 (Equations 10.23 and 10.24) are the adsorption-desorption of
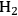 and CO, respectively. The same reaction mechanism was used to build the two models in both phases. Two additional equations were added to develop the aqueous phase model to account for the formation of gas bubbles during aqueous-phase reforming. It is reported that these models describe the experimental kinetic data reasonably well using similar values of the kinetic parameters, suggesting that the vapor phase and aqueous phase reforming chemistry is similar on platinum. The microkinetic model also suggests that the C–C bond in EG is broken on platinum at an intermediate value of x in
and CO, respectively. The same reaction mechanism was used to build the two models in both phases. Two additional equations were added to develop the aqueous phase model to account for the formation of gas bubbles during aqueous-phase reforming. It is reported that these models describe the experimental kinetic data reasonably well using similar values of the kinetic parameters, suggesting that the vapor phase and aqueous phase reforming chemistry is similar on platinum. The microkinetic model also suggests that the C–C bond in EG is broken on platinum at an intermediate value of x in
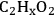 , thus supporting results from DFT calculations.
, thus supporting results from DFT calculations.
10.4.3 Changing Selectivity toward Alkanes
Until now, we have discussed the selective production of hydrogen by aqueous phase reforming of oxygenated hydrocarbons. However, it is possible to tailor aqueous phase reforming of highly oxygenated hydrocarbons, such as sorbitol, to produce a clean stream of heavier alkanes consisting of butane, pentane and hexane. This conversion requires the formation of hydrogen and
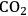 through reforming over a metal catalyst, such as Pt, as well as dehydration over a solid acid catalyst (such as silica-alumina) or with a mineral acid. Following that, the generated hydrogen is used to hydrogenate the dehydrated intermediates over the metal sites. Alkanes are produced through consecutive cycles of dehydration/hydrogenation. When the reforming, dehydration and hydrogenation steps are balanced properly, the hydrogen generated through reforming can be completely consumed for the hydrogenation of dehydrated intermediates. The alkanes formed are straight-chain compounds with only minor amounts of branched isomers (less than 5%). The selectivity toward alkanes can be modified by changing the catalyst composition, pH of the feed (when mineral acids are present), and the reaction conditions [56].
through reforming over a metal catalyst, such as Pt, as well as dehydration over a solid acid catalyst (such as silica-alumina) or with a mineral acid. Following that, the generated hydrogen is used to hydrogenate the dehydrated intermediates over the metal sites. Alkanes are produced through consecutive cycles of dehydration/hydrogenation. When the reforming, dehydration and hydrogenation steps are balanced properly, the hydrogen generated through reforming can be completely consumed for the hydrogenation of dehydrated intermediates. The alkanes formed are straight-chain compounds with only minor amounts of branched isomers (less than 5%). The selectivity toward alkanes can be modified by changing the catalyst composition, pH of the feed (when mineral acids are present), and the reaction conditions [56].
It has been reported by Huber et al. [56] that for an aqueous sorbitol feed, the
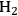 selectivity decreases from 43 to 11% for a Pt/
selectivity decreases from 43 to 11% for a Pt/
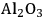 catalyst when a solid acid catalyst (
catalyst when a solid acid catalyst (
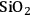 –
–
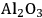 ) is added, and also decreases from 43 to 6% upon the addition of a mineral acid (HCl) to the feed to change the pH from 7 to 2. These results demonstrate that with the presence of an acid catalyst, the hydrogen generated by the reforming reaction is consumed for the production of alkanes. When 4 wt% Pt/
) is added, and also decreases from 43 to 6% upon the addition of a mineral acid (HCl) to the feed to change the pH from 7 to 2. These results demonstrate that with the presence of an acid catalyst, the hydrogen generated by the reforming reaction is consumed for the production of alkanes. When 4 wt% Pt/
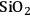 –
–
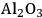 was used for the conversion of the aqueous 5 wt% sorbitol feed, the
was used for the conversion of the aqueous 5 wt% sorbitol feed, the
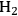 selectivity was less than 5%, indicating that most of the
selectivity was less than 5%, indicating that most of the
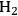 generated was consumed in the production of the alkanes. It was observed that decreasing the reaction temperature from 538 to 498 K did not have a significant effect on product selectivity. On the other hand, at 498 K, when the system pressure was increased to about 40 bar from 26 bar, the hexane selectivity was increased to 40 from 21%. The authors reported that co-feeding of
generated was consumed in the production of the alkanes. It was observed that decreasing the reaction temperature from 538 to 498 K did not have a significant effect on product selectivity. On the other hand, at 498 K, when the system pressure was increased to about 40 bar from 26 bar, the hexane selectivity was increased to 40 from 21%. The authors reported that co-feeding of
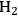 could improve the alkane production further. At 498 K and 35 bar, co-feeding of
could improve the alkane production further. At 498 K and 35 bar, co-feeding of
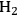 resulted in an increase in hexane selectivity to 78 from 55%. At these conditions, it was calculated that 90% of the effluent gas-phase carbon was present as alkanes. Based on these results, it can be concluded that increasing the hydrogen partial pressure in the reactor increases the rate of hydrogenation as opposed to C–C bond cleavage over metal sites. Finally, it is noteworthy that co-feeding of
resulted in an increase in hexane selectivity to 78 from 55%. At these conditions, it was calculated that 90% of the effluent gas-phase carbon was present as alkanes. Based on these results, it can be concluded that increasing the hydrogen partial pressure in the reactor increases the rate of hydrogenation as opposed to C–C bond cleavage over metal sites. Finally, it is noteworthy that co-feeding of
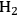 with the aqueous feed opens the possibility of using bifunctional catalysts (metal/acid) with metals (such as Pd) that by themselves show low activities for hydrogen production by APR reactions.
with the aqueous feed opens the possibility of using bifunctional catalysts (metal/acid) with metals (such as Pd) that by themselves show low activities for hydrogen production by APR reactions.
As can be seen in this section, dehydration followed by hydrogenation is a significant reaction sequence for aqueous-phase reforming to form alkanes from oxygenated hydrocarbons. For this reason, we look into the kinetics of dehydration reactions in further detail with the example of dehydration/hydrogenation of 2-butanol in aqueous environment.
Example: Dehydration/Hydrogenation of Butanol
As mentioned above, successive dehydration/hydrogenation in the aqueous phase leads to the production of straight chain alkanes such as butane, pentane and hexane from oxygenated hydrocarbons [56]. When metal catalysts such as Pt supported on acidic supports (silica-alumina or niobium phosphate) are used under conditions, such as 520 K and 50 bar, olefinic species are not observed in the outlet stream [56]. These results suggest that hydrogenation is fast and the rate limiting step is dehydration. West et al. [88] studied the kinetics of dehydration of 2-butanol in the presence of water (liquid as well as vapor) over different acid catalysts and developed a kinetic model to understand the pathways of dehydration in both phases. 2-butanol was chosen as a model compound. The authors first confirmed that dehydration was the rate limiting step by reacting 2-butanol over Pt/
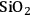 –
–
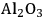 as well as the support only (
as well as the support only (
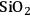 –
–
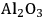 ) to convert to butane and butenes, respectively. The rates of production of butane and butenes were the same on these two catalysts, indicating that dehydration was the rate limiting step.
) to convert to butane and butenes, respectively. The rates of production of butane and butenes were the same on these two catalysts, indicating that dehydration was the rate limiting step.
The vapor pressures of water and 2-butanol at 513 K are 33 and 30 bar, respectively. With a total system pressure of 53 bar, in a closed system, water should remain as a liquid. However, when there is a gas flowing through the water, the gas bubbles carry water vapor to the outlet stream of the reactor. In addition, the butene produced by the dehydration reaction is present only in the gas phase. Therefore, the rate of butene production will also affect the extent of the sparging of liquid water. When there is no butene formation, all of the 2-butanol and water can be sparged into the gas phase at a gas flow (He) inlet to the reactor of 74 cm3(STP)/min. The extents of vaporization of water and 2-butanol were quantified by using vapor-liquid equilibrium expressions and the Antoinne equation.
Two adsorption models were used for kinetic modeling to explain the reaction kinetics data for the effect of the flow rate of inert gas on the rates of dehydration over
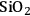 –
–
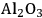 and
and
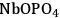 . The first mechanism investigated is a Langmuir-Hinshelwood (L-H) mechanism, which consists of the surface reaction of adsorbed butanol with a vacant site to form adsorbed butene and water. This mechanism is shown below:
. The first mechanism investigated is a Langmuir-Hinshelwood (L-H) mechanism, which consists of the surface reaction of adsorbed butanol with a vacant site to form adsorbed butene and water. This mechanism is shown below:
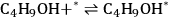
|
10.25 |
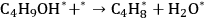
|
10.26 |
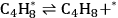
|
10.27 |
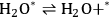
|
10.28 |
Steps 1, 3, and 4 (Equations 10.25, 10.27 and 10.28) are the adsorption-desorption steps of butanol, butene, and water, respectively, and thus are assumed to be quasi-equilibrated steps. Step 2 (Equation 10.26) is the surface reaction for the production of butene and water on the surface from adsorbed 2-butanol.
The second pathway is based on the BET adsorption isotherm, in which the possibility of multilayer water adsorption is also included. Equation 10.29 shows the fractional coverage for the first adsorbed water molecule on the surface, whereas equations 10.30 and 10.31 show the fractional coverage of two-stack and n-layer stack of water molecules, respectively.
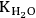 is the equilibrium constant for the adsorption of water and KW1 is the equilibrium constant for the adsorption of water on an already adsorbed water molecule. The KW1 constant is actually the equilibrium constant for liquefaction of water (inverse of the saturation pressure of water at the reaction temperature).
is the equilibrium constant for the adsorption of water and KW1 is the equilibrium constant for the adsorption of water on an already adsorbed water molecule. The KW1 constant is actually the equilibrium constant for liquefaction of water (inverse of the saturation pressure of water at the reaction temperature).
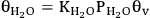
|
10.29 |
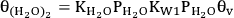
|
10.30 |
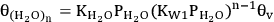
|
10.31 |
It is also possible to derive expressions for the hydration of butanol adsorbed on the surface. In such a case, the adsorption of butanol on a dry site and adsorption of water onto an adsorbed butanol molecule is added with corresponding equilibrium constants.
Figure 10.12 shows the change in butanol dehydration reaction rates in terms of TOF (1/s) with changing flow rates of the inert gas. For both solid acid catalysts (only the data for
 are shown in the figure), the general trend observed is an increase of the reaction rate at inert gas flow rates below and above 70 cm3(STP)/min approximately. Higher flow rates of inert gas increase the reaction rate by decreasing the partial pressure of water. On the other hand, with flow rates lower than 70 cm3(STP)/min, liquid water is present in the reactor and, increasing the inert gas flow rates, results in preferential vaporization of 2-butanol due to the non-ideality of the butanol-water system and high value of the butanol activity coefficient.
are shown in the figure), the general trend observed is an increase of the reaction rate at inert gas flow rates below and above 70 cm3(STP)/min approximately. Higher flow rates of inert gas increase the reaction rate by decreasing the partial pressure of water. On the other hand, with flow rates lower than 70 cm3(STP)/min, liquid water is present in the reactor and, increasing the inert gas flow rates, results in preferential vaporization of 2-butanol due to the non-ideality of the butanol-water system and high value of the butanol activity coefficient.
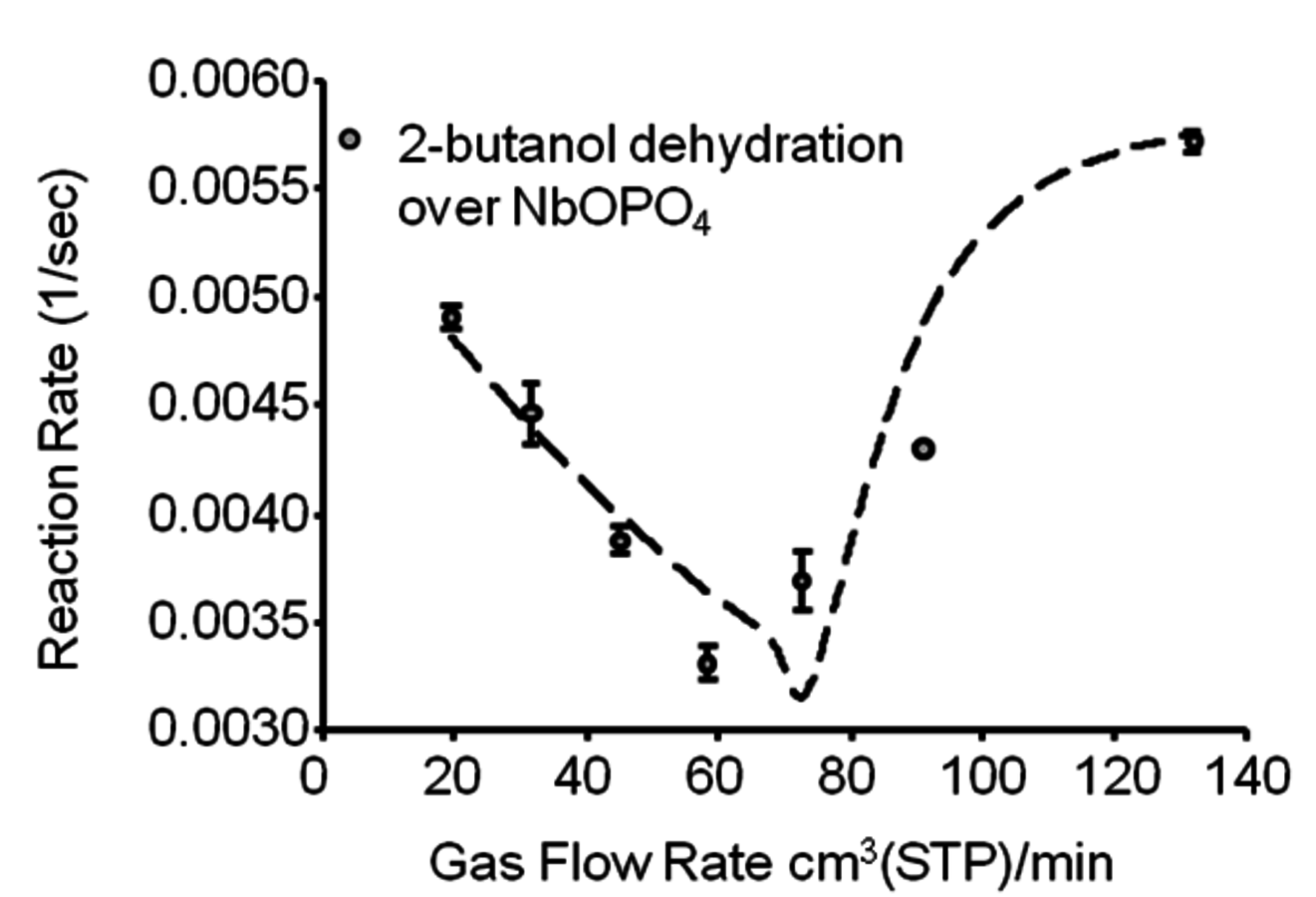
 at 513 K and 52 atm total system pressure. BET multilayer adsorption mechanism with two surface reactions (adsorbed butanol with a vacant site, and adsorbed-hydrated butanol with a hydrated site) is used for the model (shown with the dashed line). Adapted from [88]
at 513 K and 52 atm total system pressure. BET multilayer adsorption mechanism with two surface reactions (adsorbed butanol with a vacant site, and adsorbed-hydrated butanol with a hydrated site) is used for the model (shown with the dashed line). Adapted from [88]Fig. 10.12: The change in 2-butanol dehydration reaction rates in terms of TOF (1/s) with changing flow rate of the inert gas over
at 513 K and 52 atm total system pressure. BET multilayer adsorption mechanism with two surface reactions (adsorbed butanol with a vacant site, and adsorbed-hydrated butanol with a hydrated site) is used for the model (shown with the dashed line). Adapted from [88]
The simplest description of the surface reaction chemistry is the L-H mechanism in which adsorbed butanol reacts with a vacant site to produce adsorbed butene and water. However, this mechanism fails to predict accurately the behavior of the liquid/water system, because it does not include the formation of liquid water on the surface. On the other hand, for the model including the BET adsorption isotherms, a second surface reaction step can be included in which a hydrated site can also react with adsorbed-hydrated butanol to form butene and water. It can be seen in Figure 10.12, that the BET multilayer adsorption mechanism with two surface reactions (adsorbed butanol with a vacant site, and adsorbed-hydrated butanol with a hydrated site) correctly predicts the trends seen in experiments.
10.4.4 Glycerol Reforming and Fischer-Tropsch Synthesis
This section will conclude our introduction to the reforming of biomass derived oxygenated hydrocarbons. We present a process for the conversion of aqueous glycerol feeds over platinum-based catalysts to produce synthesis gas (mixture of
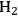 and CO) through reforming at low temperatures (498–620 K). The generated synthesis gas can subsequently be used for the production of liquid hydrocarbon fuels and/or chemicals by means of Fischer-Tropsch and methanol syntheses, respectively. This integrated process is a good example of active site coupling.
The decomposition of glycerol into CO and
and CO) through reforming at low temperatures (498–620 K). The generated synthesis gas can subsequently be used for the production of liquid hydrocarbon fuels and/or chemicals by means of Fischer-Tropsch and methanol syntheses, respectively. This integrated process is a good example of active site coupling.
The decomposition of glycerol into CO and
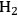 is shown by Equation 10.32, and the reforming of glycerol combined with Fischer-Tropsch reaction to form alkanes (octane in this case) is shown in Equation 10.33.
is shown by Equation 10.32, and the reforming of glycerol combined with Fischer-Tropsch reaction to form alkanes (octane in this case) is shown in Equation 10.33.
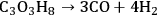
|
10.32 |
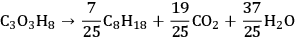
|
10.33 |
The endothermic enthalpy change of this reaction is 350 kJmol-1 and the heat generated by Fischer-Tropsch conversion of the CO and
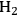 to liquid alkanes such as octane is -412 kJ mol-1. When these two reactions are combined, the overall reaction becomes exothermic with an enthalpy change of (-63 kJ mol-1). This value corresponds to about 4% of the heating value of the glycerol (-1480 kJ mol-1).
to liquid alkanes such as octane is -412 kJ mol-1. When these two reactions are combined, the overall reaction becomes exothermic with an enthalpy change of (-63 kJ mol-1). This value corresponds to about 4% of the heating value of the glycerol (-1480 kJ mol-1).
As explained earlier, APR can take place at conditions where the water-gas shift reaction is favored. Water-gas shift is an important step when the aim is to produce high yields of hydrogen in
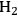 :
:
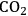 gas mixtures containing low levels of CO (~100 ppm). However, when the aim is to generate synthesis gas (
gas mixtures containing low levels of CO (~100 ppm). However, when the aim is to generate synthesis gas (
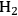 :CO mixtures), the water-gas shift reaction is an undesirable reaction. Therefore, glycerol reforming is carried out in the vapor phase at higher temperatures, where it is possible to control the extent of the water-gas shift reaction and the
:CO mixtures), the water-gas shift reaction is an undesirable reaction. Therefore, glycerol reforming is carried out in the vapor phase at higher temperatures, where it is possible to control the extent of the water-gas shift reaction and the
 :CO ratio in the outlet stream. It may also be desirable to use catalysts that do not promote the water-gas shift reaction.
:CO ratio in the outlet stream. It may also be desirable to use catalysts that do not promote the water-gas shift reaction.
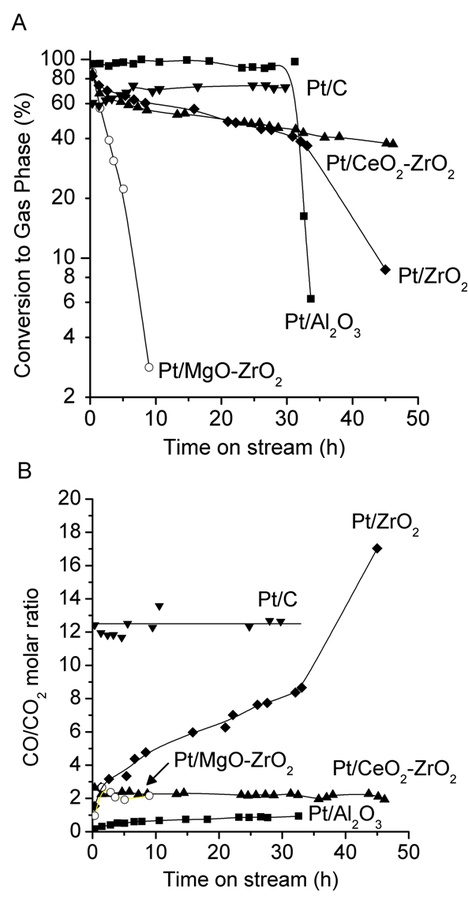
 molar ratio for catalysts consisting of Pt supported on
molar ratio for catalysts consisting of Pt supported on
 (solid squares),
(solid squares),
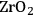 (solid diamonds),
(solid diamonds),
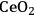 /
/
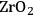 (solid triangles), MgO/
(solid triangles), MgO/
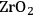 (open circles) and carbon (solid inverted triangles) with time on stream. Reactions were carried out at 623 K and 1 bar with 30% aqueous glycerol solution. Adapted from [59]
(open circles) and carbon (solid inverted triangles) with time on stream. Reactions were carried out at 623 K and 1 bar with 30% aqueous glycerol solution. Adapted from [59]Fig. 10.13: A) The conversion of glycerol to gas-phase products and B) the CO/
molar ratio for catalysts consisting of Pt supported on
(solid squares),
(solid diamonds),
/
(solid triangles), MgO/
(open circles) and carbon (solid inverted triangles) with time on stream. Reactions were carried out at 623 K and 1 bar with 30% aqueous glycerol solution. Adapted from [59]
Glycerol is a platform molecule that can be derived from various biomass conversion routes. One source of glycerol is the low value waste stream obtained from the production of biodiesel by transesterification of plant oils [53,89] and animal fat [53]. We first present studies carried out for the synthesis gas production from glycerol, and then we will discuss the combination of reforming with Fischer-Tropsch synthesis for the production of liquid fuels. Soares et al. [59] studied the gas-phase conversion of glycerol into synthesis gas over supported Pt catalysts using a feed of glycerol:
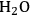 solution (30 wt% glycerol) at 623 K and atmospheric pressure. Figure 10.13 shows the conversion of glycerol and the CO/
solution (30 wt% glycerol) at 623 K and atmospheric pressure. Figure 10.13 shows the conversion of glycerol and the CO/
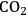 molar ratio for catalysts consisting of Pt supported on
molar ratio for catalysts consisting of Pt supported on
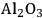 ,
,
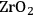 ,
,
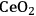 /
/
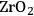 and MgO/
and MgO/
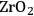 and carbon with time on stream. As it can be seen from Figure 10.13A all the catalysts except Pt/C showed deactivation with time on stream. Pt/C remained stable for at least 30 hours on stream. The different deactivation profiles suggest that the support has an important effect on the stability of the catalyst. The authors tracked the rate of formation of C2-hydrocarbons (ethane and ethylene) for the Pt supported catalysts. They recorded measurable amounts of ethane and ethylene over the Pt catalysts with oxide supports, whereas negligible amounts were detected over Pt/C. They also observed that the
and carbon with time on stream. As it can be seen from Figure 10.13A all the catalysts except Pt/C showed deactivation with time on stream. Pt/C remained stable for at least 30 hours on stream. The different deactivation profiles suggest that the support has an important effect on the stability of the catalyst. The authors tracked the rate of formation of C2-hydrocarbons (ethane and ethylene) for the Pt supported catalysts. They recorded measurable amounts of ethane and ethylene over the Pt catalysts with oxide supports, whereas negligible amounts were detected over Pt/C. They also observed that the
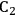 -TOF/
-TOF/
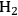 -TOF ratio increased with time on stream for the oxide supported catalysts. These observations suggest that a possible cause of deactivation is the deposition of carbonaceous species caused by unsaturated species formed through dehydration of glycerol or intermediates over oxide supports. It has also been reported that the water-gas shift activity was increased in the presence of oxide supports. As can be seen in Figure 10.13B, the initial CO/
-TOF ratio increased with time on stream for the oxide supported catalysts. These observations suggest that a possible cause of deactivation is the deposition of carbonaceous species caused by unsaturated species formed through dehydration of glycerol or intermediates over oxide supports. It has also been reported that the water-gas shift activity was increased in the presence of oxide supports. As can be seen in Figure 10.13B, the initial CO/
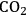 ratio for Pt/C is 12:1, whereas for the other catalysts it is less than 3:1. When the glycerol conversion is decreased by increasing space velocity or glycerol concentration, the CO/
ratio for Pt/C is 12:1, whereas for the other catalysts it is less than 3:1. When the glycerol conversion is decreased by increasing space velocity or glycerol concentration, the CO/
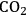 ratio is also increased, suggesting that the reforming of glycerol to CO and
ratio is also increased, suggesting that the reforming of glycerol to CO and
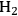 is a primary reaction, whereas the water-gas shift reaction is a secondary reaction. As a consequence of this behavior, it becomes possible to adjust the
is a primary reaction, whereas the water-gas shift reaction is a secondary reaction. As a consequence of this behavior, it becomes possible to adjust the
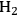 /CO ratio to be suitable for Fischer-Tropsch reaction, while reaching almost complete conversions of glycerol.
/CO ratio to be suitable for Fischer-Tropsch reaction, while reaching almost complete conversions of glycerol.
To achieve the most efficient combination of glycerol reforming with Fischer-Tropsch synthesis, the glycerol reforming step should take place at a lower temperature, such that heat integration between the endothermic reforming and exo- thermic Fischer-Tropsch reaction can be realized. Therefore, the glycerol conversion reaction was also studied at 498–573 K. As explained earlier, the conversion of polyols to
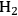 and CO requires selective C–C cleavage versus C–O bond breaking [15], and Pt is preferred for its ability to do so. However, at these low temperatures CO desorption may limit the reforming reaction due to high coverages of CO on the surface. This high coverage of CO on the surface may also increase the rate of the water-gas shift reaction, which is not a desirable reaction when the purpose is to generate synthesis gas.
and CO requires selective C–C cleavage versus C–O bond breaking [15], and Pt is preferred for its ability to do so. However, at these low temperatures CO desorption may limit the reforming reaction due to high coverages of CO on the surface. This high coverage of CO on the surface may also increase the rate of the water-gas shift reaction, which is not a desirable reaction when the purpose is to generate synthesis gas.
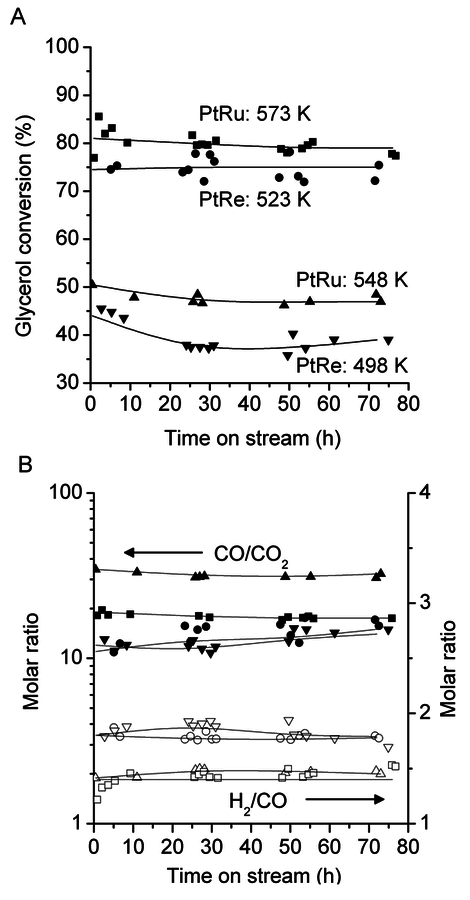
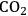 (solid symbols) and
(solid symbols) and
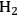 /CO (open symbols) with time on stream at low temperatures over PtRu and PtRe (atomic ratio 1:1) bimetallic catalysts. Reactions were carried out at 1 bar with 30% aqueous glycerol solution. Triangles: PtRu/C, 548 K, squares: PtRu/C, 573 K, inverted triangles: PtRe/C, 498 K, and circles: PtRe/C 523 K. Adapted from [59]
/CO (open symbols) with time on stream at low temperatures over PtRu and PtRe (atomic ratio 1:1) bimetallic catalysts. Reactions were carried out at 1 bar with 30% aqueous glycerol solution. Triangles: PtRu/C, 548 K, squares: PtRu/C, 573 K, inverted triangles: PtRe/C, 498 K, and circles: PtRe/C 523 K. Adapted from [59]Fig. 10.14: A) The glycerol conversion to gas-phase products and B) molar ratios of CO/
(solid symbols) and
/CO (open symbols) with time on stream at low temperatures over PtRu and PtRe (atomic ratio 1:1) bimetallic catalysts. Reactions were carried out at 1 bar with 30% aqueous glycerol solution. Triangles: PtRu/C, 548 K, squares: PtRu/C, 573 K, inverted triangles: PtRe/C, 498 K, and circles: PtRe/C 523 K. Adapted from [59]
Therefore, it is crucial to find a catalyst that is still active and selective for C–C cleavage, and that can weaken the CO bonding to catalyst surface. The binding energy of CO can be reduced by using appropriate metal alloys, such as PtRu and PtRe alloy catalysts [90]. Figure 10.14 shows the glycerol conversion and molar ratios of CO/
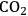 and
and
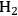 /CO with time on stream at low temperatures over PtRu and PtRe (atomic ratio 1:1) bimetallic catalysts.
/CO with time on stream at low temperatures over PtRu and PtRe (atomic ratio 1:1) bimetallic catalysts.
These results demonstrate that the conversion of glycerol to synthesis gas can be accomplished at temperatures within the ranges used for Fischer-Tropsch synthesis [91]. It is important to note that the
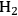 /CO ratio can be adjusted to be suitable for Fischer-Tropsch synthesis (
/CO ratio can be adjusted to be suitable for Fischer-Tropsch synthesis (
 /CO=2 stoichiometrically) if necessary through a water-gas shift reaction. Over 10 wt% PtRe/C (atomic ratio 1:1), higher pressures and feed concentrations were studied to allow for an efficient combination of two processes. Simonetti et al. [58] investigated the formation of liquid alkanes by the integration of glycerol conversion with Fischer-Tropsch synthesis. These experiments employed a two-bed catalyst system using 1.0 g of 10 wt% PtRe (1:1)/C followed by 1.7 g of 1.0 wt% Ru/
/CO=2 stoichiometrically) if necessary through a water-gas shift reaction. Over 10 wt% PtRe/C (atomic ratio 1:1), higher pressures and feed concentrations were studied to allow for an efficient combination of two processes. Simonetti et al. [58] investigated the formation of liquid alkanes by the integration of glycerol conversion with Fischer-Tropsch synthesis. These experiments employed a two-bed catalyst system using 1.0 g of 10 wt% PtRe (1:1)/C followed by 1.7 g of 1.0 wt% Ru/
 , with an 80 wt% glycerol feed at 548 K and total pressures between 5 and 17 bar. Figure 10.15 demonstrates the coupling of glycerol reforming and Fischer-Tropsch synthesis to produce liquid fuels, and Figure 10.16 shows the results of the experiments carried out at different pressures. At 5 bar, only 32% of the carbon is converted to alkanes and the primary product is observed to be CO. When pressure is increased to 11 and 17 bar, the carbon distribution is shifted toward C1–C5+ alkanes, such that selectivities of alkanes are 42% and 51% at 11 bar and 17 bar, respectively. In addition to the C5+ alkanes, the organic liquid effluent also contains oxygenates, such as acetone, pentanones, hexanones, and heptanones. The amount of carbon in these oxygenates increases by a factor of 5 with increasing pressure. At the highest pressure studied, the amount of CO decreased by more than an order of magnitude and total selectivity to alkanes increased. On the other hand, compared to 11 bar, selectivity toward C5+ alkanes is slightly reduced. This change is due to increased water-gas shift activity as well as overall Fischer-Tropsch activity with increased pressure. The percentage of carbon in the organic liquid-phase product was 43% at 17 bar, 35% at 11 bar, and 15% at 5 bar, with the percentage of carbon in gaseous products (CO,
, with an 80 wt% glycerol feed at 548 K and total pressures between 5 and 17 bar. Figure 10.15 demonstrates the coupling of glycerol reforming and Fischer-Tropsch synthesis to produce liquid fuels, and Figure 10.16 shows the results of the experiments carried out at different pressures. At 5 bar, only 32% of the carbon is converted to alkanes and the primary product is observed to be CO. When pressure is increased to 11 and 17 bar, the carbon distribution is shifted toward C1–C5+ alkanes, such that selectivities of alkanes are 42% and 51% at 11 bar and 17 bar, respectively. In addition to the C5+ alkanes, the organic liquid effluent also contains oxygenates, such as acetone, pentanones, hexanones, and heptanones. The amount of carbon in these oxygenates increases by a factor of 5 with increasing pressure. At the highest pressure studied, the amount of CO decreased by more than an order of magnitude and total selectivity to alkanes increased. On the other hand, compared to 11 bar, selectivity toward C5+ alkanes is slightly reduced. This change is due to increased water-gas shift activity as well as overall Fischer-Tropsch activity with increased pressure. The percentage of carbon in the organic liquid-phase product was 43% at 17 bar, 35% at 11 bar, and 15% at 5 bar, with the percentage of carbon in gaseous products (CO,
 , and C1–C9 alkanes) decreasing from 71% at 5 bar to approximately 50% at 11 and 17 bar. At 5 and 11 bar, 14% of the carbon is contained as oxygenated species in the aqueous effluent (acetone, methanol, and ethanol), and at 17 bar, this value decreases to 10%. These aqueous liquid effluents contain between 5 wt% and 15 wt% methanol, ethanol, and acetone. It is noted that these species are at similar concentrations as aqueous ethanol streams produced by fermentation of glucose (e.g., 5 wt%). Therefore, it might be desirable to separate the components by distillation for use in the chemical industry.
, and C1–C9 alkanes) decreasing from 71% at 5 bar to approximately 50% at 11 and 17 bar. At 5 and 11 bar, 14% of the carbon is contained as oxygenated species in the aqueous effluent (acetone, methanol, and ethanol), and at 17 bar, this value decreases to 10%. These aqueous liquid effluents contain between 5 wt% and 15 wt% methanol, ethanol, and acetone. It is noted that these species are at similar concentrations as aqueous ethanol streams produced by fermentation of glucose (e.g., 5 wt%). Therefore, it might be desirable to separate the components by distillation for use in the chemical industry.
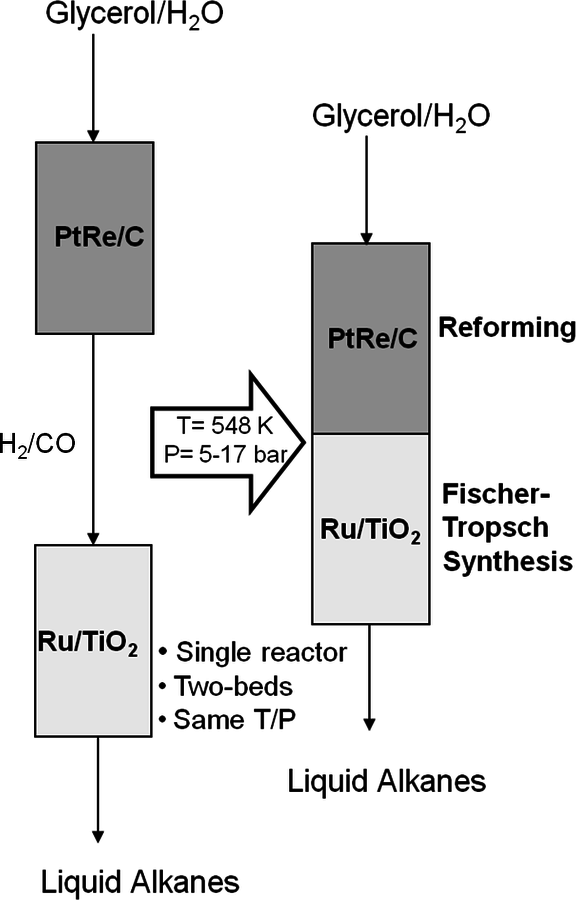
Fig. 10.15: Schematic representation of the dual-bed catalytic reactor used for the combined glycerol reforming and Fischer-Tropsch synthesis to produce liquid alkanes
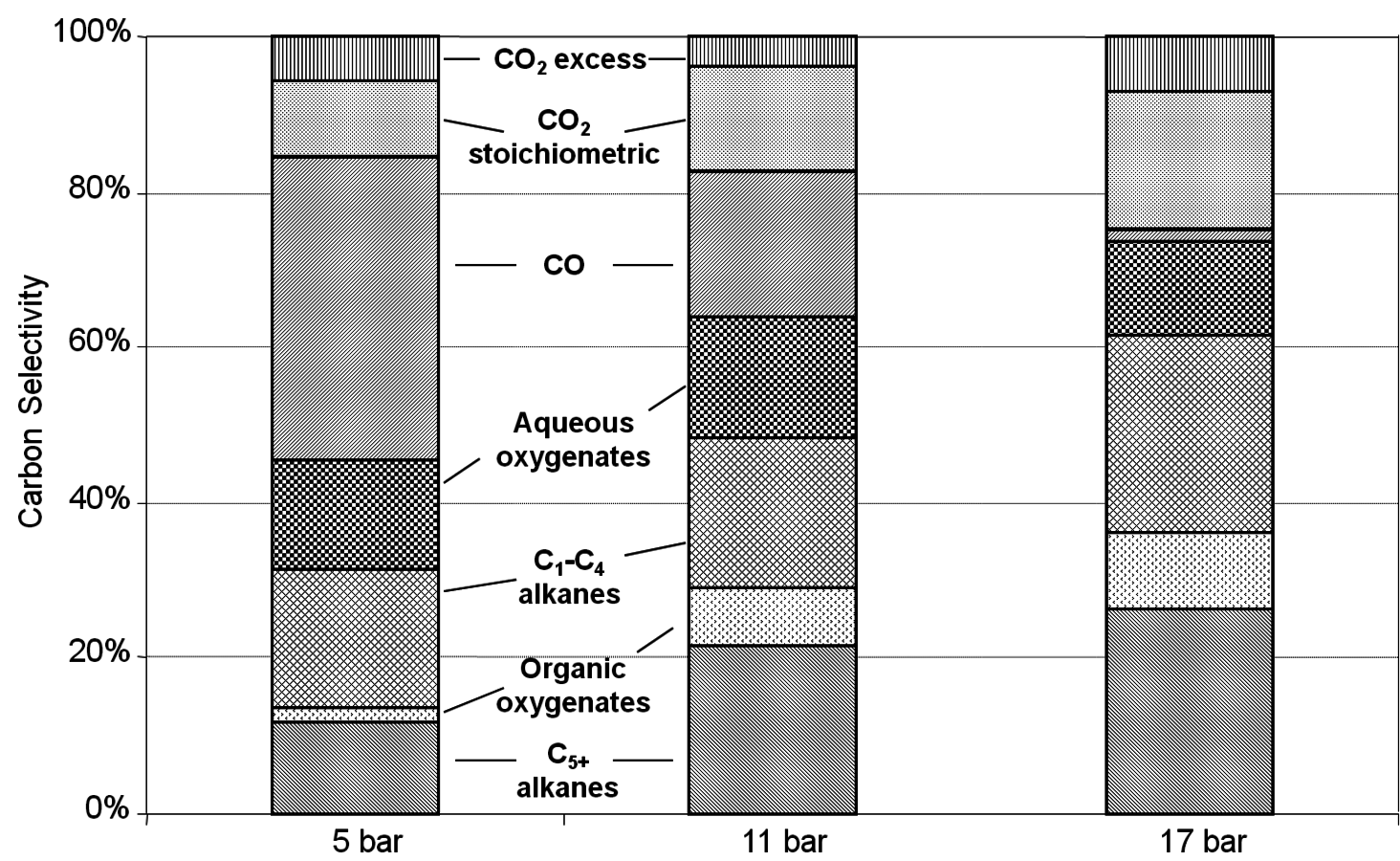
 at 548 K at pressures of 5, 11 and 17 bar. Adapted from [58]
at 548 K at pressures of 5, 11 and 17 bar. Adapted from [58]Fig. 10.16: Carbon distribution in the effluent from glycerol conversion over a two-bed catalytic reactor containing PtRe/C followed by Ru/
at 548 K at pressures of 5, 11 and 17 bar. Adapted from [58]
10.5 Conversion of Sugars/Polyols to Liquid Fuels via the Formation of Monofunctional Intermediates
Until now, we have presented the utilization of aqueous-phase or gas-phase reforming reactions to form
 , synthesis gas (
, synthesis gas (
 and CO) or alkanes (C4–C6) through selective C–C or C–O cleavage steps. Even when the alkanes are obtained selectively, the carbon number of the heaviest alkane is limited to the carbon number of the sugar or polyol feed. As we concluded in the previous sections, the production of heavier liquid fuels from sugars and/or polyols can be accomplished by controlled oxygen removal reactions (controlled C–O and C–C cleavage reactions) to obtain functional intermediates that can undergo C–C bond forming reactions. Kunkes et al. [14] recently reported a process in which sugars and polyols are converted to monofunctional intermediates over a carbon-supported PtRe bimetallic catalyst in a single reactor. By operating at moderate temperatures (283–523 K) and pressures (20–30 bar) with concentrated aqueous polyol (60 wt% sorbitol) or sugar (40 wt% glucose) feeds, 80% of the initial oxygen content of the sugars and polyols is removed by controlling C–C cleavage (leading to
and CO) or alkanes (C4–C6) through selective C–C or C–O cleavage steps. Even when the alkanes are obtained selectively, the carbon number of the heaviest alkane is limited to the carbon number of the sugar or polyol feed. As we concluded in the previous sections, the production of heavier liquid fuels from sugars and/or polyols can be accomplished by controlled oxygen removal reactions (controlled C–O and C–C cleavage reactions) to obtain functional intermediates that can undergo C–C bond forming reactions. Kunkes et al. [14] recently reported a process in which sugars and polyols are converted to monofunctional intermediates over a carbon-supported PtRe bimetallic catalyst in a single reactor. By operating at moderate temperatures (283–523 K) and pressures (20–30 bar) with concentrated aqueous polyol (60 wt% sorbitol) or sugar (40 wt% glucose) feeds, 80% of the initial oxygen content of the sugars and polyols is removed by controlling C–C cleavage (leading to
 and
and
 ) and C–O cleavage (leading to alkanes) rates. As a result, an organic phase is formed that spontaneously separates from water and consists of a mixture of C4–C6 monofunctional species (carboxylic acids, alcohols, ketones and heterocyclic species). Cleavage of C–O bonds takes place through hydrogenolysis, which is promoted by Re [92,93]. As oxygen is progressively removed from the intermediates, the binding of these intermediates to the catalyst surface becomes weaker, resulting in the production of monofunctional species, such as acids, alcohols, ketones and heterocycles before alkanes are generated. One of the most significant features of this process is that the deoxygenation (C–O bond cleavage) is accomplished by using the hydrogen generated in situ by the endothermic partial reforming of the carbohydrate feed. The exothermic deoxygenation reactions are balanced with endothermic reforming reactions in the same reactor, such that the overall conversion is mildly exothermic and more than 90% of the energy content of the carbohydrate feed is stored in the reaction products. The overall process is illustrated schematically for sorbitol in Figure 10.17.
) and C–O cleavage (leading to alkanes) rates. As a result, an organic phase is formed that spontaneously separates from water and consists of a mixture of C4–C6 monofunctional species (carboxylic acids, alcohols, ketones and heterocyclic species). Cleavage of C–O bonds takes place through hydrogenolysis, which is promoted by Re [92,93]. As oxygen is progressively removed from the intermediates, the binding of these intermediates to the catalyst surface becomes weaker, resulting in the production of monofunctional species, such as acids, alcohols, ketones and heterocycles before alkanes are generated. One of the most significant features of this process is that the deoxygenation (C–O bond cleavage) is accomplished by using the hydrogen generated in situ by the endothermic partial reforming of the carbohydrate feed. The exothermic deoxygenation reactions are balanced with endothermic reforming reactions in the same reactor, such that the overall conversion is mildly exothermic and more than 90% of the energy content of the carbohydrate feed is stored in the reaction products. The overall process is illustrated schematically for sorbitol in Figure 10.17.
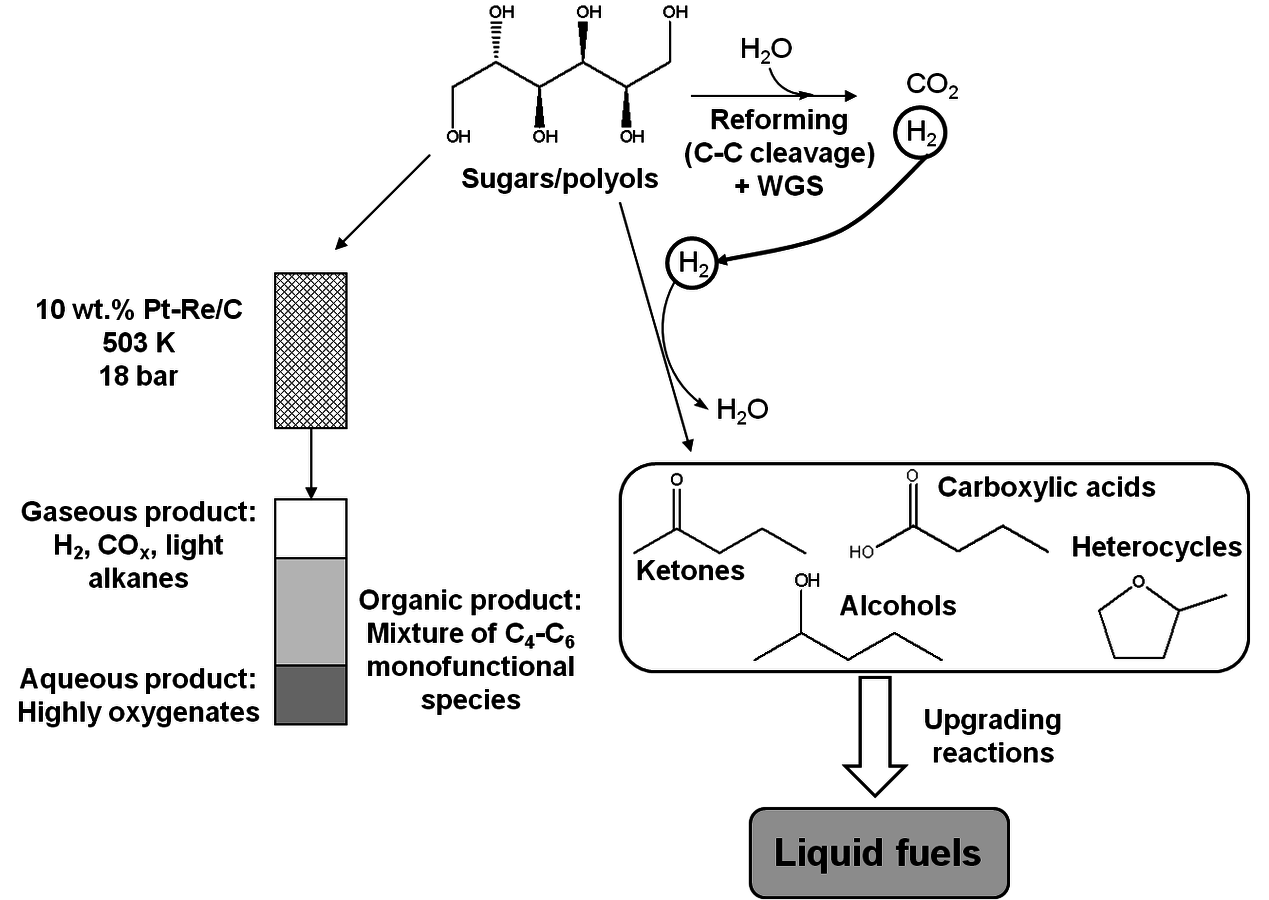
Fig. 10.17: Schematic representation of the conversion of sugars and polyols over PtRe/C to produce an organic liquid consisting of C4–C6 monofunctional species (ketones, alcohols, carboxylic acids and heterocyclic species). Adapted from [14]
The gas-phase effluent consists of
 and light alkanes (C1–C6), the aqueous phase consists of higher oxygenates such as isosorbide, and the organic phase effluent consists of the monofunctional species and alkanes (C4–C6). The conversion of a 60 wt% sorbitol solution over a 10 wt% PtRe(1:1) catalyst supported on carbon was studied in a fixed bed flow reactor at pressures of 18–27 bar, temperatures of 483–523 K, and weight hourly space velocities (WHSV) of 0.6 to 2.4 hr-1. The effluent carbon from the aqueous phase species is shifted to organic phase species when pressure is increased from 18 to 27 bar at 483 K. At a higher temperature (503 K), the pressure increase from 18 to 27 bar results in a shift from aqueous phase species to gaseous species, whereas it is observed that pressure has a negligible effect on the carbon distribution, when the temperature is increased further to 523 K. The production of alkanes increases at the expense of oxygenated species as pressure or temperature is increased when the other is kept constant. At constant temperature and pressure, when the space velocity is increased from 0.60 to 1.2 h-1, the production of organic phase species, specifically ketones, alcohols and carboxylic acids, increases at the expense of alkanes in the gaseous phase as well as in the liquid organic phase. However, a further increase of space velocity to 2.4 h-1 shifts the carbon distribution toward the aqueous phase oxygenates. At 503 K and 18 bar, with WHSV of 0.6 h-1, PtRe/C showed good stability for longer than one month time on stream. The organic outlet stream at these conditions contains 52% of the carbon found in the 60 wt% sorbitol feed, when the gaseous and aqueous streams contain 36% and 12%, respectively.
and light alkanes (C1–C6), the aqueous phase consists of higher oxygenates such as isosorbide, and the organic phase effluent consists of the monofunctional species and alkanes (C4–C6). The conversion of a 60 wt% sorbitol solution over a 10 wt% PtRe(1:1) catalyst supported on carbon was studied in a fixed bed flow reactor at pressures of 18–27 bar, temperatures of 483–523 K, and weight hourly space velocities (WHSV) of 0.6 to 2.4 hr-1. The effluent carbon from the aqueous phase species is shifted to organic phase species when pressure is increased from 18 to 27 bar at 483 K. At a higher temperature (503 K), the pressure increase from 18 to 27 bar results in a shift from aqueous phase species to gaseous species, whereas it is observed that pressure has a negligible effect on the carbon distribution, when the temperature is increased further to 523 K. The production of alkanes increases at the expense of oxygenated species as pressure or temperature is increased when the other is kept constant. At constant temperature and pressure, when the space velocity is increased from 0.60 to 1.2 h-1, the production of organic phase species, specifically ketones, alcohols and carboxylic acids, increases at the expense of alkanes in the gaseous phase as well as in the liquid organic phase. However, a further increase of space velocity to 2.4 h-1 shifts the carbon distribution toward the aqueous phase oxygenates. At 503 K and 18 bar, with WHSV of 0.6 h-1, PtRe/C showed good stability for longer than one month time on stream. The organic outlet stream at these conditions contains 52% of the carbon found in the 60 wt% sorbitol feed, when the gaseous and aqueous streams contain 36% and 12%, respectively.
Kunkes et al. [14] also showed that glucose can be converted directly to mixtures of monofunctional species without the need for initial formation of sorbitol through a hydrogenation step. An aqueous solution containing 40 wt% glucose was used with slightly different reaction conditions (483 K, 18 bar, and 0.6 hr-1 WHSV) to ensure stable catalyst activity. The most significant difference in product distributions compared to sorbitol conversion is the higher selectivity towards carboxylic acids. Carboxylic acids constitute 14% of total carbon, or 35% of organic product from glucose conversion as opposed to 6% of the total carbon, or 13% of the organic product from sorbitol conversion.
10.5.1 Upgrading Strategies for Monofunctional Intermediates to Obtain Liquid Fuels
As stated earlier, the monofunctional species obtained in the organic effluent from the conversion of sugars and polyols over PtRe/C catalyst have sufficient functional moieties to undergo various upgrading reactions to form high-molecular weight compounds suitable for gasoline, diesel and jet-fuel applications. These upgrading strategies are summarized in Figure 10.18. Diesel fuels consist primarily of hydrocarbons with a linear carbon chain and minimal branching to achieve high cetane numbers, whereas gasoline typically contains more highly branched hydrocarbons and aromatic compounds having high octane numbers [94,95].
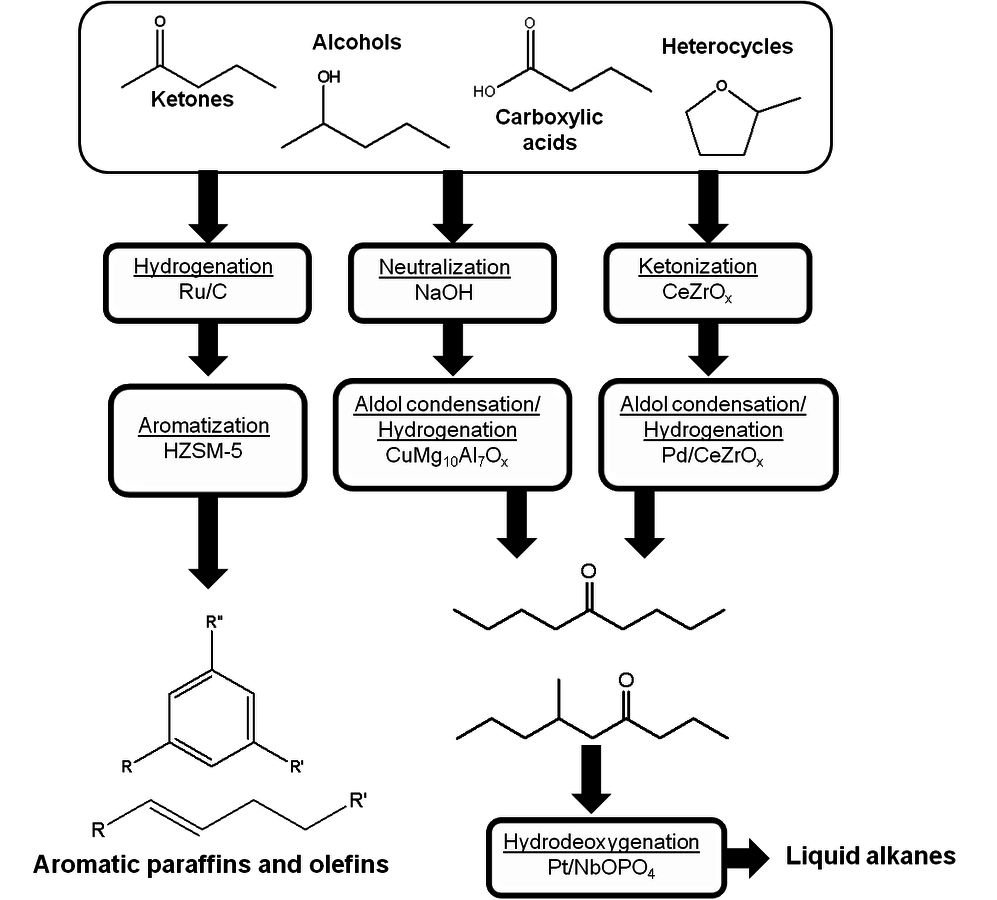
Fig. 10.18: Upgrading strategies for the production of liquid fuels starting from monofunctional species obtained from sugars/polyols over PtRe/C. Adapted from [14]
With respect to gasoline components, Kunkes et al. [14] showed that the organic liquid produced from sorbitol can be converted to aromatic compounds or to a distribution of branched olefins centered at C12. The first step for both conversions is the hydrogenation of ketones and carboxylic acids to alcohols. Using a 5 wt% Ru/C catalyst at 433 K, under 55 bar
 , most of the ketones, carboxylic acids and some of the heterocyclic species in the sorbitol-derived organic liquid can be hydrogenated into corresponding alcohols. It is important to note that the heterocycles present in the organic liquid can be used as high-octane additives in gasoline [94,96,97]. In order to obtain aromatic species, the mixture of alcohols is reacted over HZSM-5 at 673 K and atmospheric pressure [98]. When hydrogenation is combined with the aromatization step, 25% and 29% of the carbon in the sorbitol-derived organic phase is converted to paraffins and olefins containing 3 and 4 carbon atoms, respectively. Almost 40% of the inlet carbon is converted to aromatic species, consisting of benzene, toluene,
, most of the ketones, carboxylic acids and some of the heterocyclic species in the sorbitol-derived organic liquid can be hydrogenated into corresponding alcohols. It is important to note that the heterocycles present in the organic liquid can be used as high-octane additives in gasoline [94,96,97]. In order to obtain aromatic species, the mixture of alcohols is reacted over HZSM-5 at 673 K and atmospheric pressure [98]. When hydrogenation is combined with the aromatization step, 25% and 29% of the carbon in the sorbitol-derived organic phase is converted to paraffins and olefins containing 3 and 4 carbon atoms, respectively. Almost 40% of the inlet carbon is converted to aromatic species, consisting of benzene, toluene,
 -benzene (a benzene with two additional carbon atom substituents such as xylenes or ethyl benzene) and C3–C6 substituted benzene. In addition, it is possible to dehydrate secondary alcohols obtained by hydrogenation over an acidic niobia catalyst to form
-benzene (a benzene with two additional carbon atom substituents such as xylenes or ethyl benzene) and C3–C6 substituted benzene. In addition, it is possible to dehydrate secondary alcohols obtained by hydrogenation over an acidic niobia catalyst to form
 –
–
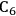 branched olefins. These olefins can later undergo an oligomerization reaction combined with cracking reactions over HZSM-5 to produce a distribution of branched olefins centered at C12. Approximately 50% of the carbon present in the secondary alcohols can be converted into fuel-grade compounds overall [14].
branched olefins. These olefins can later undergo an oligomerization reaction combined with cracking reactions over HZSM-5 to produce a distribution of branched olefins centered at C12. Approximately 50% of the carbon present in the secondary alcohols can be converted into fuel-grade compounds overall [14].
To produce C8–C12 compounds that have minimal branching to be used in diesel fuel applications, C–C coupling by aldol condensation can be applied to the organic effluent obtained from sorbitol solution over PtRe/C. However, when this organic liquid is subjected to aldol condensation/hydrogenation in the presence of
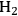 over a bifunctional CuMg10Al7Ox catalyst (has both base and metal sites), it was observed that the small amounts of carboxylic acids and esters caused deactivation of the basic CuMg10Al7Ox catalyst [99]. Therefore, the esters were hydrolyzed and the acids were neutralized by refluxing the organic mixture with a 20 wt% NaOH solution at 343 K. Following this step, the aldol condensation/hydrogenation of the
over a bifunctional CuMg10Al7Ox catalyst (has both base and metal sites), it was observed that the small amounts of carboxylic acids and esters caused deactivation of the basic CuMg10Al7Ox catalyst [99]. Therefore, the esters were hydrolyzed and the acids were neutralized by refluxing the organic mixture with a 20 wt% NaOH solution at 343 K. Following this step, the aldol condensation/hydrogenation of the
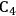 –
–
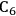 ketones and secondary alcohols was carried out over CuMg10Al7Ox at 573 K and 5 bar pressure with
ketones and secondary alcohols was carried out over CuMg10Al7Ox at 573 K and 5 bar pressure with
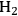 co-feed [55,57]. 45 % of the inlet carbon was converted to
co-feed [55,57]. 45 % of the inlet carbon was converted to
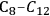 species containing 1 or no oxygen atoms. The remaining carbon is in the form of light species (C4–C6) with 1 or no oxygen atoms. The 3-ketones showed low reactivity for aldol condensation [100], while heterocyclic species were mostly inert. The product stream of the aldol condensation step can be subjected to hydrodeoxygenation over a metal/acid bifunctional catalyst to form an alkane stream [19].
species containing 1 or no oxygen atoms. The remaining carbon is in the form of light species (C4–C6) with 1 or no oxygen atoms. The 3-ketones showed low reactivity for aldol condensation [100], while heterocyclic species were mostly inert. The product stream of the aldol condensation step can be subjected to hydrodeoxygenation over a metal/acid bifunctional catalyst to form an alkane stream [19].
As an alternative to the neutralization/ester hydrolysis step (NaOH refluxing), the carboxylic acid molecules can be upgraded by coupling through ketonization reactions to form higher molecular weight ketones (releasing
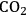 and
and
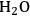 ), prior to the aldol condensation step. Ketonization reactions are advantageous especially for the organic liquids obtained from glucose, since the carboxylic acid production is significant. Ketonization of the monofunctional mixture obtained from glucose over CeZrOx achieved greater than 98% conversion of the carboxylic acids in the feed to
), prior to the aldol condensation step. Ketonization reactions are advantageous especially for the organic liquids obtained from glucose, since the carboxylic acid production is significant. Ketonization of the monofunctional mixture obtained from glucose over CeZrOx achieved greater than 98% conversion of the carboxylic acids in the feed to
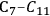 ketones. During the ketonization reaction, the remainder of the monofunctional species remains unreacted, and therefore, the product stream of this step can be subjected to further C–C coupling by aldol condensation of ketones and secondary alcohols. Even though the CuMg10Al7Ox catalyst still deactivated due to trace amounts of organic acids and esters, a bifunctional catalyst consisting of 0.25 wt% Pd on CeZrOx was stable at 623 K with time on stream for aldol condensation. The feed used for aldol condensation over the Pd/CeZrOx catalyst consisted of 34%
ketones. During the ketonization reaction, the remainder of the monofunctional species remains unreacted, and therefore, the product stream of this step can be subjected to further C–C coupling by aldol condensation of ketones and secondary alcohols. Even though the CuMg10Al7Ox catalyst still deactivated due to trace amounts of organic acids and esters, a bifunctional catalyst consisting of 0.25 wt% Pd on CeZrOx was stable at 623 K with time on stream for aldol condensation. The feed used for aldol condensation over the Pd/CeZrOx catalyst consisted of 34%
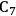 –C11 ketones and 66% C4–C6 monofunctionals and alkanes. When the ketonized organic liquid was reacted over Pd/CeZrOx at 573 K and 5 bar pressure with
–C11 ketones and 66% C4–C6 monofunctionals and alkanes. When the ketonized organic liquid was reacted over Pd/CeZrOx at 573 K and 5 bar pressure with
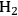 co-feed, 57% of the total carbon in the liquid organic product stream was in the form of C7+ ketones, with 34% and 23% generated in ketonization and aldol condensation reactions, respectively. Products with a carbon chain length greater than C12 were also observed, due to aldol condensation of
co-feed, 57% of the total carbon in the liquid organic product stream was in the form of C7+ ketones, with 34% and 23% generated in ketonization and aldol condensation reactions, respectively. Products with a carbon chain length greater than C12 were also observed, due to aldol condensation of
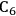 ketones with C7+ ketones formed during ketonization [14].
ketones with C7+ ketones formed during ketonization [14].
10.5.2 Integration of C–C Coupling Reactions of Monofunctional Intermediates
As mentioned earlier, ketonization is an effective strategy for decreasing the acidity of the aldol condensation/hydrogenation feed, allowing for the integration of the two steps using a basic aldol condensation catalyst. Since the reaction conditions for ketonization and aldol condensation/hydrogenation reactions are similar, it is possible that they could be integrated in a single reactor with a dual-bed system consisting of CeZrOx to perform ketonization followed by a downstream bed of Pd/CeZrOx to perform aldol condensation/hydrogenation. Integrating the aldol condensation and ketonization steps into a single reactor system is an example of active site coupling that would streamline the overall C–C coupling process. To effectively accomplish the integration of ketonization and aldol condensation/hydrogenation in a single reactor, it is necessary to understand the effects of the co-products, i.e.,
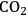 and water, produced during the ketonization of carboxylic acids on the downstream aldol condensation/hydrogenation reaction. To study the effect of
and water, produced during the ketonization of carboxylic acids on the downstream aldol condensation/hydrogenation reaction. To study the effect of
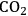 and water on aldol condensation activity, experiments with pure 2-hexanone as the feed were carried out. When 10 mol%
and water on aldol condensation activity, experiments with pure 2-hexanone as the feed were carried out. When 10 mol%
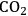 in
in
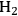 stream was co-fed with 2-hexanone feed over 0.25 wt% Pd/CeZrOx, 2-hexanone self-condensation conversion decreased from 60% to 5%. Since the basic sites are poisoned by
stream was co-fed with 2-hexanone feed over 0.25 wt% Pd/CeZrOx, 2-hexanone self-condensation conversion decreased from 60% to 5%. Since the basic sites are poisoned by
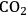 , the reactions that can take place on acid and metal sites become more pronounced, causing higher yields to alkanes through dehydration/hydrogenation and reforming reactions. When water was introduced with a 2-butanone feed (12 wt%), a 40% decrease in self-condensation conversion was recorded [101].
, the reactions that can take place on acid and metal sites become more pronounced, causing higher yields to alkanes through dehydration/hydrogenation and reforming reactions. When water was introduced with a 2-butanone feed (12 wt%), a 40% decrease in self-condensation conversion was recorded [101].
Literature shows that the nature of the interaction of
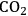 with a mixed oxide surface can be altered by changing the composition of mixed oxides [102,103]. To permit the integration of ketonization and aldol condensation reactions in a single reactor,
with a mixed oxide surface can be altered by changing the composition of mixed oxides [102,103]. To permit the integration of ketonization and aldol condensation reactions in a single reactor,
 mixed oxide catalysts with different compositions were synthesized to find a catalyst, over which inhibition by
mixed oxide catalysts with different compositions were synthesized to find a catalyst, over which inhibition by
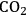 and
and
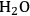 was minimal [104]. It was observed that the conversion of 2-hexanone increases with increasing zirconia content in mixed oxide catalysts and pure
was minimal [104]. It was observed that the conversion of 2-hexanone increases with increasing zirconia content in mixed oxide catalysts and pure
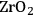 displays the highest conversion of 90%. When 10%
displays the highest conversion of 90%. When 10%
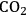 in
in
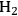 co-feed was introduced instead of a pure
co-feed was introduced instead of a pure
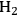 stream, all ceria containing catalysts lost significant activity. On the other hand,
stream, all ceria containing catalysts lost significant activity. On the other hand,
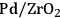 showed significant resistance to inhibition by
showed significant resistance to inhibition by
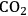 , with only a 20% decrease in condensation activity (corresponding to 72% conversion of 2-hexanone). In addition, when water was introduced in a 2-butanone feed, the catalytic activity decreased by about 10% for the case of
, with only a 20% decrease in condensation activity (corresponding to 72% conversion of 2-hexanone). In addition, when water was introduced in a 2-butanone feed, the catalytic activity decreased by about 10% for the case of
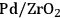 compared to a 40% decrease for Pd/CeZrOx. Thus, it was concluded that
compared to a 40% decrease for Pd/CeZrOx. Thus, it was concluded that
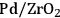 was a suitable catalyst for integrating ketonization and aldol condensation/hydrogenation in a single reactor due to the diminished inhibition of aldol condensation activity by water and
was a suitable catalyst for integrating ketonization and aldol condensation/hydrogenation in a single reactor due to the diminished inhibition of aldol condensation activity by water and
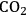 . This conclusion was then tested over a dual-catalyst bed system consisting of CeZrOx (upstream bed for ketonization) and
. This conclusion was then tested over a dual-catalyst bed system consisting of CeZrOx (upstream bed for ketonization) and
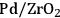 (downstream bed for aldol condensation/hydrogenation) using a feed mixture of 20 mol% butanoic acid in 2-hexanone in comparison to a feed mixture of 20 mol% heptane in 2-hexanone at 623 K and 5 bar with
(downstream bed for aldol condensation/hydrogenation) using a feed mixture of 20 mol% butanoic acid in 2-hexanone in comparison to a feed mixture of 20 mol% heptane in 2-hexanone at 623 K and 5 bar with
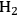 co-feed. The difference between the aldol condensation activity and selectivity values for the two runs were found to be insignificant, suggesting that
co-feed. The difference between the aldol condensation activity and selectivity values for the two runs were found to be insignificant, suggesting that
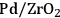 showed resistance to the
showed resistance to the
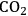 and water generated in the ketonization step when butanoic acid was present in the feed [104].
and water generated in the ketonization step when butanoic acid was present in the feed [104].
Following these results, the dual-catalyst bed system was implemented to process the organic liquid of monofunctionals obtained from a 60 wt% aqueous sorbitol solution over PtRe/C. Specifically, the performance of the dual-bed system was compared to that of a cascade system, in which two separate reactors are used for ketonization CeZrOx) and aldol condensation/hydrogenation (
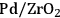 ), with removal of
), with removal of
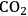 and water between the reactors [12]. The reactor set-ups are shown in Figure 10.19. The overall conversion of monofunctional species was approximately 70% for both systems, with a 42% overall yield to C7+ products. Accordingly, the selectivity to C7+ products was approximately 60%, the remainder of the products being in the form of C4–C6 products. Effluent streams from both set-ups can be subjected to dehydration/hydrogenation reactions over Pt/SiO2–Al2O3 to obtain alkane streams. It was concluded in this work that the dual-bed, single reactor system is the preferred mode of operation, because the energy consumption as well as the reactor infrastructure associated with cooling and re-heating the products in between two C–C coupling steps can be eliminated, decreasing operating and capital costs [12].
and water between the reactors [12]. The reactor set-ups are shown in Figure 10.19. The overall conversion of monofunctional species was approximately 70% for both systems, with a 42% overall yield to C7+ products. Accordingly, the selectivity to C7+ products was approximately 60%, the remainder of the products being in the form of C4–C6 products. Effluent streams from both set-ups can be subjected to dehydration/hydrogenation reactions over Pt/SiO2–Al2O3 to obtain alkane streams. It was concluded in this work that the dual-bed, single reactor system is the preferred mode of operation, because the energy consumption as well as the reactor infrastructure associated with cooling and re-heating the products in between two C–C coupling steps can be eliminated, decreasing operating and capital costs [12].
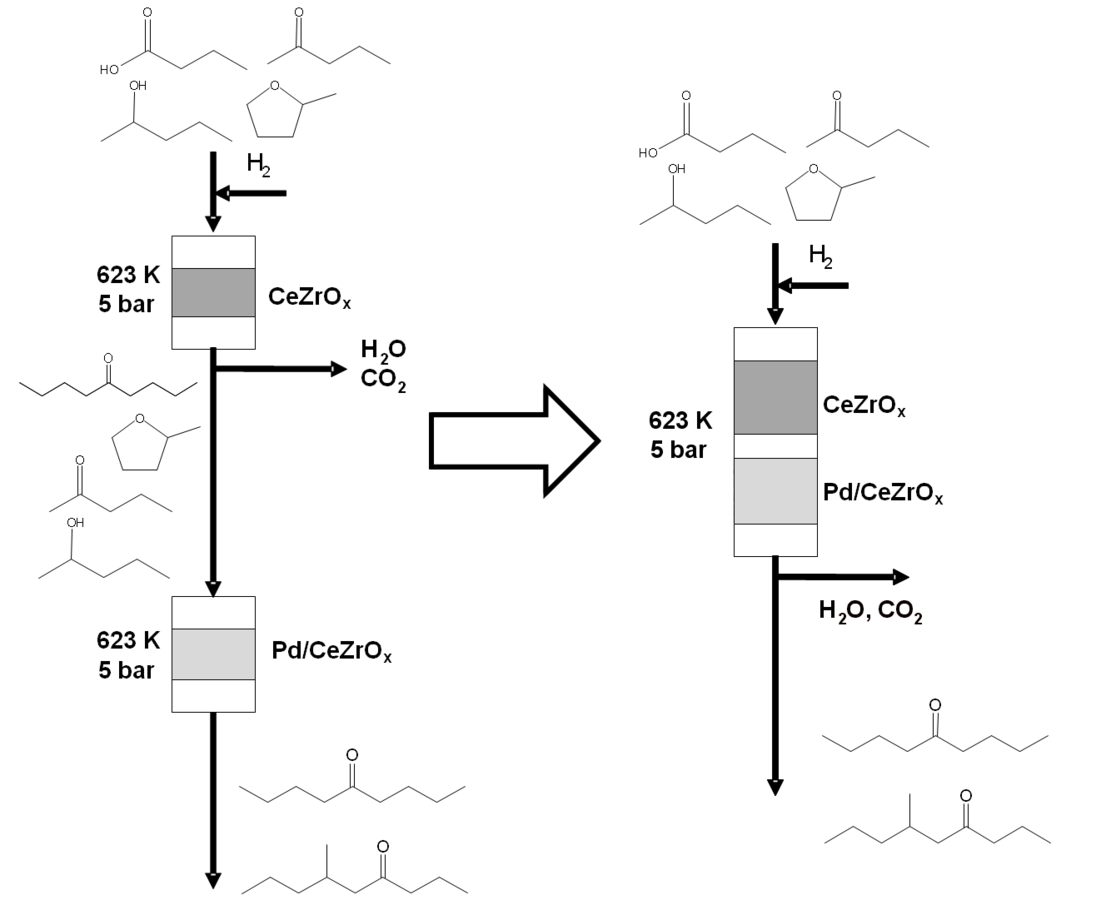
Fig. 10.19: Schematic representation of the dual-bed catalytic reactor used for the combined ketonization and aldol condensation to produce liquid alkanes starting from monofunctional species obtained from sugars/polyols over PtRe/C. Adapted from [12]
In the final product stream from both set-ups, the ratio of linear alkanes to branched alkanes was determined to be approximately equal to 1. The majority of the branched ketones have minimal branching, i.e. methyl-branched species, which are formed from the coupling of 2-ketones and/or secondary alcohols. Only small amounts of ethyl-branched alkanes were observed, suggesting that coupling of 2-ketones/alcohols with 3-ketones/alcohols is not as favorable. The linear alkanes are formed by coupling of 2-ketones/alcohols with aldehydes generated from ring-opening of heterocycles over
 . These results show that C–C coupling through ketonization and aldol condensation is desirable for the production of diesel fuel, since the extent of branching in the long carbon chain alkanes is minimal and can be controlled [12].
. These results show that C–C coupling through ketonization and aldol condensation is desirable for the production of diesel fuel, since the extent of branching in the long carbon chain alkanes is minimal and can be controlled [12].
10.6 Levulinic Acid and ɣ-Valerolactone Platforms for the Production of Liquid Fuels
Levulinic acid (LA) is an attractive platform molecule from which a range of fine chemicals (δ-aminolevulinic acid, diphenolic acid, etc.) and fuel additives (levulinate esters, methyltetrahydrofuran, etc.) can be produced [105]. Levulinic acid can be obtained from the decomposition of 5-hydroxymethylfurfural (HMF) or direct hydrolysis of lignocellulosic biomass [106,107] and cellulose [108] through dilute sulfuric acid hydrolysis. In all of these conversions, equimolar amounts of formic acid are generated with levulinic acid. Levulinic acid production by hydrolysis of cellulose can be carried out through different strategies, such as by using dilute sulfuric acid [108], concentrated hydrochloric acid [109], solid acids [110] and ionic liquids [111]. Among these strategies, hydrolysis by dilute sulfuric acid appears to have the optimum balance of cost, yield, and scalability for the preparation of levulinic acid. The Biofine process produces levulinic acid from lignocellulose at a pilot level production via dilute sulfuric acid hydrolysis [106,107]. Final yields to levulinic acid are around 70–80% of the theoretical maximum, and correspond to 50% of the mass of the C6 sugars. The remaining mass is collected as formic acid (20%), which is separated from levulinic acid by evaporation, and humins (30%), which are solid polymers produced by degradation reactions of [112].
A specifically valuable derivative of LA is ɣ-valerolactone (GVL) [112], another platform molecule, from which fine chemicals, fuel additives, and gasoline, jet fuel and diesel fuel components can be produced [10,108,113,114]. LA can be converted to GVL through consecutive dehydration/hydrogenation reactions. If dehydration takes place first, angelica lactone is the intermediate, and if reduction takes place first, 4-hydroxy-pentanoic acid is the intermediate. The conversion of LA to GVL can take place at relatively low temperatures (373–543 K) and high pressures (50–150 bars) with either homogeneous or heterogeneous catalysts [115]. High yields of GVL (97%) can be obtained using a Ru/C catalyst. The reduction of LA to GVL generally takes place in the presence of molecular
 ; however, the use of
; however, the use of
 generated in situ from the decomposition of formic acid is a promising alternative. The use of formic acid as a hydrogen source in aqueous solutions also eliminates the need for purification of LA. When obtained through mineral acid hydrolysis from cellulose, the separation of levulinic acid from the mineral acid becomes energy intensive. A recent strategy for the production of GVL starting from cellulose, starts with the hydrolysis of cellulose with dilute sulfuric acid solution to generate an equimolar mixture of levulinic and formic acids in water. Levulinic acid is then reduced to GVL over a Ru/C catalyst using the
generated in situ from the decomposition of formic acid is a promising alternative. The use of formic acid as a hydrogen source in aqueous solutions also eliminates the need for purification of LA. When obtained through mineral acid hydrolysis from cellulose, the separation of levulinic acid from the mineral acid becomes energy intensive. A recent strategy for the production of GVL starting from cellulose, starts with the hydrolysis of cellulose with dilute sulfuric acid solution to generate an equimolar mixture of levulinic and formic acids in water. Levulinic acid is then reduced to GVL over a Ru/C catalyst using the
 generated in situ by formic acid decomposition. Even though sulfuric acid inhibits the reduction activity significantly, the Ru/C shows stable activity. The GVL product is more hydrophobic than levulinic acid, and therefore, selective separation of GVL from sulfuric acid becomes possible using a hydrophobic extracting solvent. This way, most of the sulfuric acid can be recycled back to the cellulose deconstruction reactor. For example, using an equal mass of ethyl acetate with aqueous GVL solution, 76% of the GVL can be extracted, while only 3% of the sulfuric acid and 6% of water is transferred into the organic solvent [108].
generated in situ by formic acid decomposition. Even though sulfuric acid inhibits the reduction activity significantly, the Ru/C shows stable activity. The GVL product is more hydrophobic than levulinic acid, and therefore, selective separation of GVL from sulfuric acid becomes possible using a hydrophobic extracting solvent. This way, most of the sulfuric acid can be recycled back to the cellulose deconstruction reactor. For example, using an equal mass of ethyl acetate with aqueous GVL solution, 76% of the GVL can be extracted, while only 3% of the sulfuric acid and 6% of water is transferred into the organic solvent [108].
In terms of chemicals, some compounds that can be produced from GVL are α-methylene-ɣ-valerolactone [116], caprolactone [117] or adipic acid [117]. In terms of fuel applications, GVL itself can be directly used as a fuel additive (i.e., it has a similar capacity to ethanol [118]) or can be converted to methyltetrahydrofuran. In addition to forming fuel additives, two recent strategies have been reported in which GVL can be converted to liquid fuels in the range of diesel and jet fuel. In the first strategy, GVL is converted over bifunctional metal/acid catalysts (e.g., Pd/Nb2O5) to form pentanoic acid, which is used to produce 5-nonanone by ketonization at high yields (92%) [114]. 5-Nonanone can undergo hydrodeoxygenation to nonane for use in diesel fuel blends, or it can be hydrogenated/dehydrated to produce nonene, which can be converted to C18 olefins through oligomerization reactions [114,119]. In the second strategy, GVL is converted to butene through decarboxylation over an acidic catalyst, such as
 . Butene can be oligomerized to obtain jet fuel range olefins [10]. These strategies derived from GVL are summarized in Figure 10.20, which includes the production of GVL from levulinic acid. We now explore these two strategies in further detail.
. Butene can be oligomerized to obtain jet fuel range olefins [10]. These strategies derived from GVL are summarized in Figure 10.20, which includes the production of GVL from levulinic acid. We now explore these two strategies in further detail.
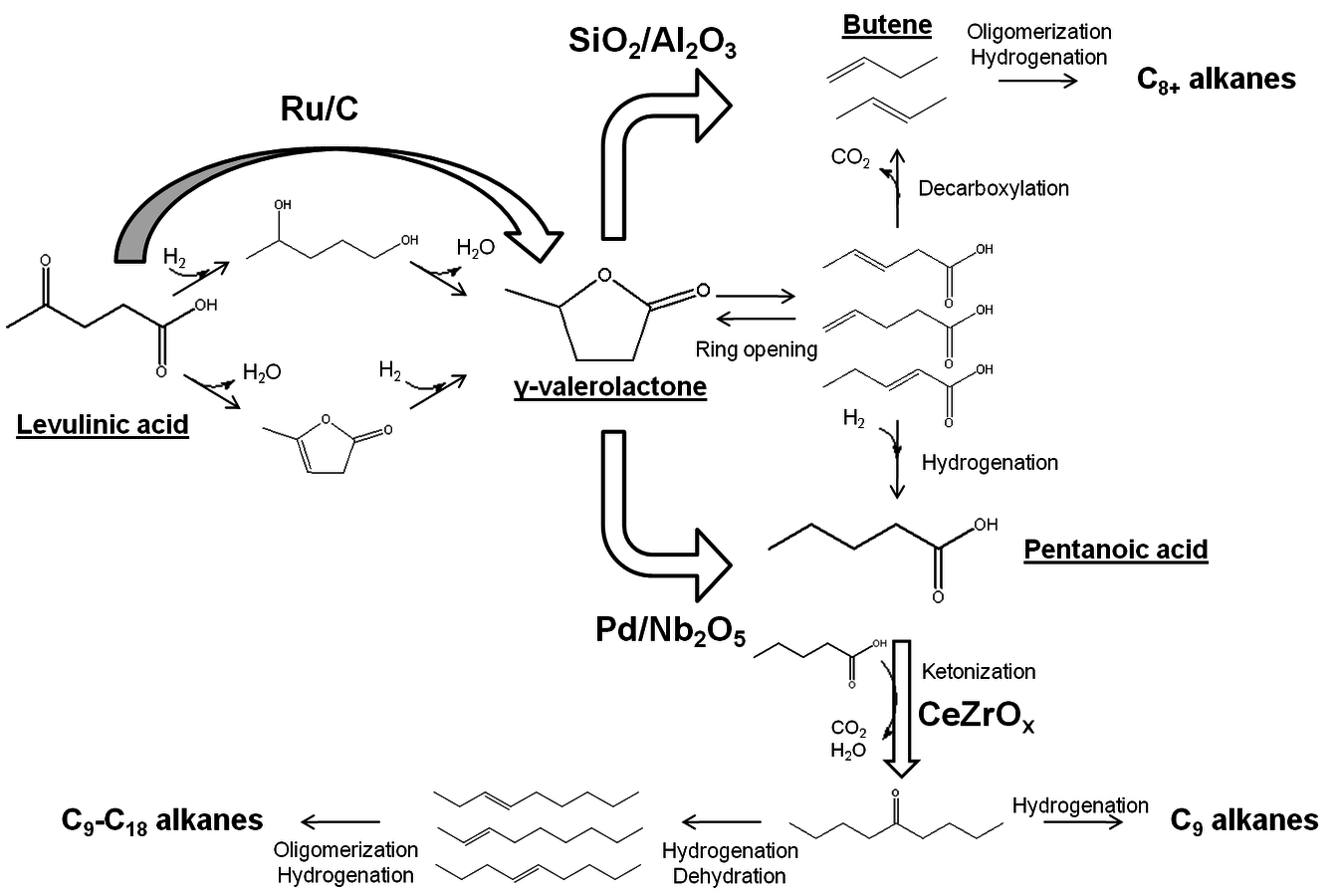
Fig. 10.20: Reaction pathways for the production of γ-valerolactone (GVL) derived from levulinic acid and the production of targeted molecular weight alkanes via the catalytic upgrading of GVL through the formation of pentanoic acid and butene isomers. Adapted from [119]
The production of pentanoic acid from aqueous solutions of GVL (50 wt%) over Pd/
 takes place at 623 K and under 35 bar with
takes place at 623 K and under 35 bar with
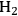 co-feed and goes through pentenoic acid intermediates obtained by ring-opening of GVL [114]. Pentenoic acids can be hydrogenated to form pentanoic acid over the metal sites (Pd) in the presence of
co-feed and goes through pentenoic acid intermediates obtained by ring-opening of GVL [114]. Pentenoic acids can be hydrogenated to form pentanoic acid over the metal sites (Pd) in the presence of
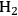 . Due to its low solubility in water, pentanoic acid can be obtained as an organic layer that spontaneously separates from water. The metal loading of the catalyst is crucial to obtain pentanoic acid with high selectivity. When the metal loading is increased, the production of byproducts, such as butane and pentane, is increased. Butane is formed through decarboxylation/decarbonylation of pentanoic acid followed by hydrogenation, and pentane can be obtained by hydrogenation/dehydration/hydrogenation of the pentanoic acid with the use of both acid and metal sites. Increasing hydrogen pressure has the same effect as increasing the metal loading. Approximately 92% yield to pentanoic acid can be achieved when the metal loading is 0.1 wt% and the
. Due to its low solubility in water, pentanoic acid can be obtained as an organic layer that spontaneously separates from water. The metal loading of the catalyst is crucial to obtain pentanoic acid with high selectivity. When the metal loading is increased, the production of byproducts, such as butane and pentane, is increased. Butane is formed through decarboxylation/decarbonylation of pentanoic acid followed by hydrogenation, and pentane can be obtained by hydrogenation/dehydration/hydrogenation of the pentanoic acid with the use of both acid and metal sites. Increasing hydrogen pressure has the same effect as increasing the metal loading. Approximately 92% yield to pentanoic acid can be achieved when the metal loading is 0.1 wt% and the
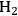 partial pressure is 17 bar (35 bar of total pressure) at 598 K. Pentanoic acid is a monofunctional molecule and can further be upgraded selectively through C–C coupling reactions. Similar to the acids obtained from sorbitol/glucose over PtRe/C bimetallic catalyst, pentanoic acid can be ketonized over a
partial pressure is 17 bar (35 bar of total pressure) at 598 K. Pentanoic acid is a monofunctional molecule and can further be upgraded selectively through C–C coupling reactions. Similar to the acids obtained from sorbitol/glucose over PtRe/C bimetallic catalyst, pentanoic acid can be ketonized over a
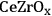 catalyst at 698 K and pressures from 1 to 20 bar to form 5-nonanone, CO2 and water. When reaction conditions for both steps are optimized for highest yields, 84% overall yield to 5-nonanone can be obtained in a cascade mode, with a 6% yield to lower molecular weight ketones, i.e., 2-hexanone and 3-heptanone [114]. These latter ketones are obtained by α and β scissions of 5-nonanone. As noted above, the 5-nonanone can be a precursor for fuels suitable for gasoline and diesel fuel.
catalyst at 698 K and pressures from 1 to 20 bar to form 5-nonanone, CO2 and water. When reaction conditions for both steps are optimized for highest yields, 84% overall yield to 5-nonanone can be obtained in a cascade mode, with a 6% yield to lower molecular weight ketones, i.e., 2-hexanone and 3-heptanone [114]. These latter ketones are obtained by α and β scissions of 5-nonanone. As noted above, the 5-nonanone can be a precursor for fuels suitable for gasoline and diesel fuel.
When GVL is reacted over an acid catalyst, like
 without the presence of metal sites, decarboxylation of GVL is the most favorable reaction, leading to butene and
without the presence of metal sites, decarboxylation of GVL is the most favorable reaction, leading to butene and
 as the main products. High yields of butene can be achieved from aqueous solutions (30–60 wt%) of GVL at 648 K and pressures ranging from 1 to 36 bar [10]. When 80% GVL in water is used as the feed at 648 K and 36 bar, a 99% conversion of GVL and 96% yield of butene can be obtained. As mentioned earlier, butene can be oligomerized to obtain high molecular weight olefins suitable for jet fuel. However, before going into details of that reaction, we present studies on thermodynamics and kinetics involved in the production of pentenoic acid, butene/
as the main products. High yields of butene can be achieved from aqueous solutions (30–60 wt%) of GVL at 648 K and pressures ranging from 1 to 36 bar [10]. When 80% GVL in water is used as the feed at 648 K and 36 bar, a 99% conversion of GVL and 96% yield of butene can be obtained. As mentioned earlier, butene can be oligomerized to obtain high molecular weight olefins suitable for jet fuel. However, before going into details of that reaction, we present studies on thermodynamics and kinetics involved in the production of pentenoic acid, butene/
 , and pentanoic acid from GVL.
, and pentanoic acid from GVL.
Fig. 10.21: Values of ΔG° and ΔH° (kJ mol-1) for γ-valerolactone (GVL) ring-opening reaction to isomers of pentenoic acid, and decarboxylation and hydrogenation of pentenoic acids to butene and pentanoic acid, respectively. Adapted from [120]
GVL conversion is initiated by ring-opening over acid sites to form an isomeric mixture of pentenoic acids. These unsaturated acids can be hydrogenated over metal sites to form pentanoic acid or can be decarboxylated over acid sites to form butene and
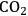 . Bond et al. [120] carried out thermodynamic calculations at standard conditions, and values of ΔG° and ΔH° for GVL ring-opening reactions to various isomers of pentenoic acid, and decarboxylation and hydrogenation of pentenoic acids are shown in Figure 10.21.
. Bond et al. [120] carried out thermodynamic calculations at standard conditions, and values of ΔG° and ΔH° for GVL ring-opening reactions to various isomers of pentenoic acid, and decarboxylation and hydrogenation of pentenoic acids are shown in Figure 10.21.
Pentenoic acid isomers consist of 4-, 2-trans, 3-trans, 2-cis, and 3-cis pentenoic acids. The enthalpy changes for the production of 4-, 2-trans, 3-trans, 2-cis, and 3-cis pentenoic acids are estimated to be 36, 17, 25, 26, 32 kJ/mol, respectively. Similarly, changes in Gibbs free energy at 298 K are estimated to be 25, 8, 14, 16, and 19 kJ/mol for 4-, 2-trans, 3-trans, 2-cis, and 3-cis pentenoic acids, respectively. These values indicate that formation of GVL is thermodynamically
 production at different weight hourly space velocities (WHSV, h-1) and temperatures (K) for GVL conversion over
production at different weight hourly space velocities (WHSV, h-1) and temperatures (K) for GVL conversion over
 . Solid inverted triangles: 498 K, open squares: 523 K, solid circles: 548 K, solid triangles: 573 K, open circles: 598 K, and solid squares: 623 K. Adapted from [120]
. Solid inverted triangles: 498 K, open squares: 523 K, solid circles: 548 K, solid triangles: 573 K, open circles: 598 K, and solid squares: 623 K. Adapted from [120]Fig. 10.22: A) Measured ratio of pentenoic acids (PEA) to γ-valerolactone (GVL) in the outlet stream and B) the rate of
production at different weight hourly space velocities (WHSV, h-1) and temperatures (K) for GVL conversion over
. Solid inverted triangles: 498 K, open squares: 523 K, solid circles: 548 K, solid triangles: 573 K, open circles: 598 K, and solid squares: 623 K. Adapted from [120]
favored over pentenoic acid isomers at standard conditions. However, since the ring-opening reaction is endothermic and entropically favored, the ratio of pentenoic acid isomers to GVL is expected to increase with temperature. This behavior is demonstrated in Figure 10.22A. As shown in Figure 10.21, hydrogenation of pentenoic acid (using an average distribution of isomers) to pentanoic acid is thermodynamically favorable with an enthalpy change of -111 kJ/mol and a Gibbs free energy change of -75 kJ/mol. This conversion makes the overall conversion of GVL to pentanoic acid favorable, with values of ΔH° and ΔG° for reaction being -84 and -59 kJ/mol, respectively. Decarboxylation of pentenoic acid to butene is also favorable, with a value of ΔG° being -93 kJ/mol. Starting from GVL, ΔG° is equal to -77 kJ/mol. Thus, the conversions of both GVL and pentenoic to butene and
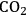 are essentially irreversible.
are essentially irreversible.
In terms of the kinetics of ring-opening and decarboxylation of GVL, Figure 10.22 shows the values of the ratio of pentenoic acid to GVL, and the
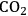 production rate with changing weight hourly space velocity (WHSV) and temperature [120]. As temperature increases from 523 K to 623 K, the pentenoic acid (PEA)/GVL ratio increases as mentioned earlier. At a constant temperature, the PEA/GVL ratio remains constant for low to moderate space velocities, indicating the presence of an equilibrium controlled reaction. At high space velocities, the ratio begins to decrease, because the rate of ring-opening becomes slower relative to the inlet feed rate. Figure 10.22B shows that the
production rate with changing weight hourly space velocity (WHSV) and temperature [120]. As temperature increases from 523 K to 623 K, the pentenoic acid (PEA)/GVL ratio increases as mentioned earlier. At a constant temperature, the PEA/GVL ratio remains constant for low to moderate space velocities, indicating the presence of an equilibrium controlled reaction. At high space velocities, the ratio begins to decrease, because the rate of ring-opening becomes slower relative to the inlet feed rate. Figure 10.22B shows that the
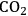 production rate is increased as temperature is increased. At a constant temperature, when the space velocity is changed, the
production rate is increased as temperature is increased. At a constant temperature, when the space velocity is changed, the
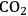 production rate does not seem to be affected. This behavior results because the conversion of GVL to pentenoic acid isomers is quasi-equilibrated and the partial pressures of GVL and pentenoic acid remain approximately at the same ratio. The decarboxylation step is kinetically controlled; therefore, the production rate of
production rate does not seem to be affected. This behavior results because the conversion of GVL to pentenoic acid isomers is quasi-equilibrated and the partial pressures of GVL and pentenoic acid remain approximately at the same ratio. The decarboxylation step is kinetically controlled; therefore, the production rate of
 remains the same with constant partial pressure of pentenoic acid and GVL [120].
remains the same with constant partial pressure of pentenoic acid and GVL [120].
Bond et al. [121] in a different work investigated the significance of the direct pathway of decarboxylation of GVL to form butene, as opposed to the indirect pathway, in which pentenoic acid isomers are generated prior to butene formation. The overall reaction scheme is shown in Figure 10.23. The relative rates of the direct and indirect decarboxylation pathways were studied by measuring the production rate of butene and
 together with the concentration of pentenoic acids with changing weight hourly space velocity. Using a 10 wt% GVL solution in water as the feed, the partial pressure of pentenoic acid continuously decreases with increasing space velocity and extrapolates to zero at infinite WHSV (1/WHSV=0). Similarly, the partial pressure of butene approaches to zero when 1/WHSV approaches to zero, whereas the GVL partial pressure approaches to that of the inlet feed. As the space velocity increases, the rate of decarboxylation remains constant and starts to decrease at lower space times (< 0.01h). This behavior suggests that the ring-opening reaction to pentenoic acids is quasi-equilibrated at relatively low space velocities, and the rate of decarboxylation is controlled by direct and indirect rates of decarboxylation. In the limit of zero space time, when there is no pentenoic acid, the rate of decarboxylation can be extrapolated to a finite number, proving the presence of a direct pathway from GVL. This value of the rate of decarboxylation at the limit of infinite space velocity is equal to the rate of direct decarboxylation of GVL to butene, and at higher space times when the indirect pathway also becomes significant, the direct pathway contributes considerably to the total butene production rate.
together with the concentration of pentenoic acids with changing weight hourly space velocity. Using a 10 wt% GVL solution in water as the feed, the partial pressure of pentenoic acid continuously decreases with increasing space velocity and extrapolates to zero at infinite WHSV (1/WHSV=0). Similarly, the partial pressure of butene approaches to zero when 1/WHSV approaches to zero, whereas the GVL partial pressure approaches to that of the inlet feed. As the space velocity increases, the rate of decarboxylation remains constant and starts to decrease at lower space times (< 0.01h). This behavior suggests that the ring-opening reaction to pentenoic acids is quasi-equilibrated at relatively low space velocities, and the rate of decarboxylation is controlled by direct and indirect rates of decarboxylation. In the limit of zero space time, when there is no pentenoic acid, the rate of decarboxylation can be extrapolated to a finite number, proving the presence of a direct pathway from GVL. This value of the rate of decarboxylation at the limit of infinite space velocity is equal to the rate of direct decarboxylation of GVL to butene, and at higher space times when the indirect pathway also becomes significant, the direct pathway contributes considerably to the total butene production rate.
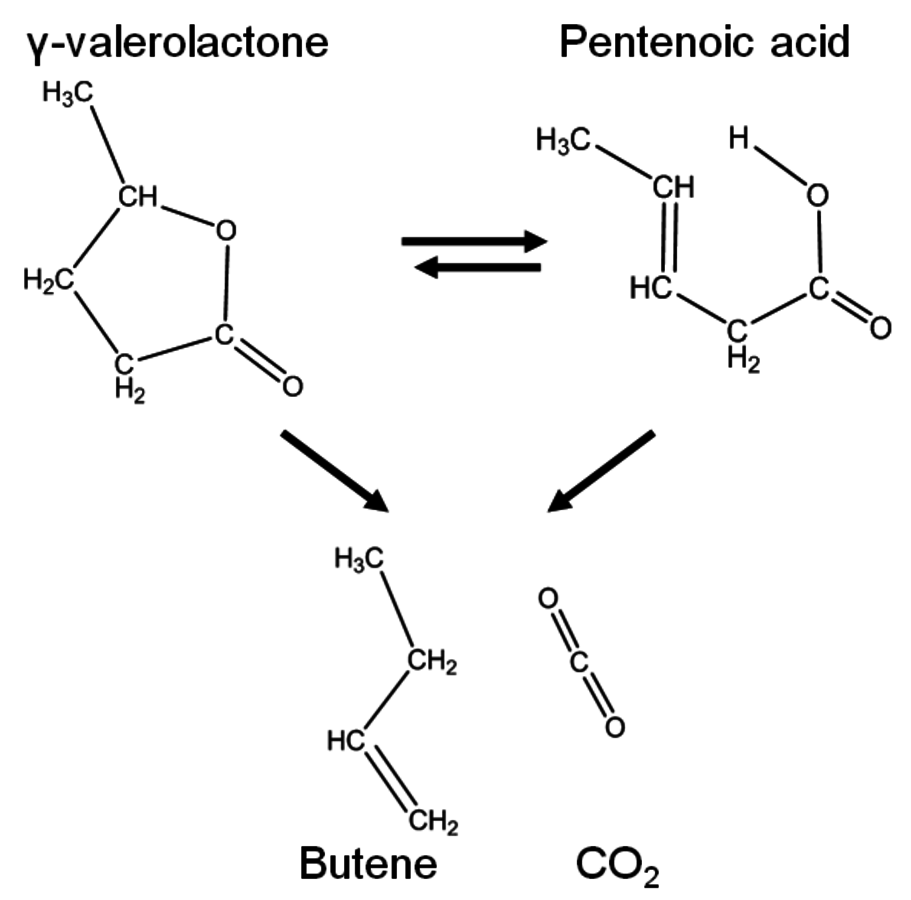
 from γ-valerolactone (GVL). Adapted from [121]
from γ-valerolactone (GVL). Adapted from [121]Fig. 10.23: The reaction schemes for the indirect and direct pathways for the production of butene/
from γ-valerolactone (GVL). Adapted from [121]
Following the formation of butene, in a second reactor connected in series, butene molecules can undergo C–C coupling reactions through oligomerization over an acid catalyst to form C8+ alkenes that can be used as jet fuel upon hydrogenation. Good yields were obtained from butene oligomerization at elevated pressures (35 bar) over Amberlyst 70. It was presented by Bond et al. [10] that the two reactors could operate at the same system pressure and therefore could be integrated in series. In the integrated system, the outlet of the first reactor is used as the feed for the second reactor. Because
 and water are present in the outlet stream with butene from the first reactor, the effects of these species on the oligomerization activity over Amberlyst 70 were investigated.
and water are present in the outlet stream with butene from the first reactor, the effects of these species on the oligomerization activity over Amberlyst 70 were investigated.
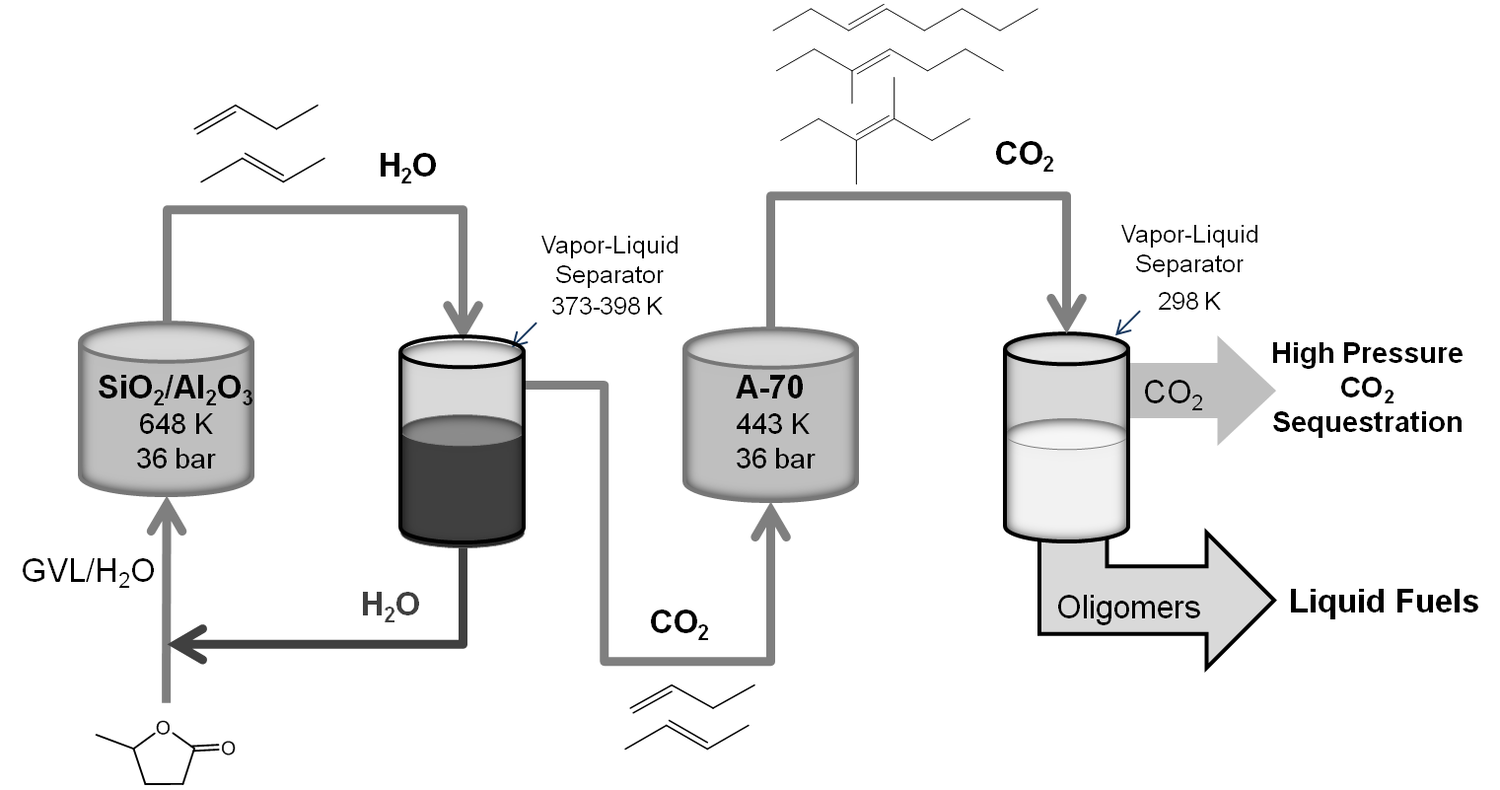
Fig. 10.24: The overall process scheme for the production of liquid fuels starting from aqueous solutions of γ-valerolactone (GVL). Adapted from [10]
Experiments showed that the presence of
 in the butene stream only affects the oligomerization reaction due to the dilution of the reactant (butene). On the other hand, when water was introduced to the inlet stream, a significant inhibition of the oligomerization activity was observed. In fact, for an equal molar feed of butene with
in the butene stream only affects the oligomerization reaction due to the dilution of the reactant (butene). On the other hand, when water was introduced to the inlet stream, a significant inhibition of the oligomerization activity was observed. In fact, for an equal molar feed of butene with
 and water, the catalytic activity was completely suppressed. The negative effect of water required that it be removed from the gas stream prior to the oligomerization reaction. This removal was achieved by using a liquid-vapor separator operated at system pressure (36 bar) prior to the oligomerization reactor. The temperature of the separator was adjusted (373–393 K) such that most of the water would be condensed (97%) with all of the butene remaining in the vapor phase. When decarboxylation of GVL and oligomerization of butene take place over
and water, the catalytic activity was completely suppressed. The negative effect of water required that it be removed from the gas stream prior to the oligomerization reaction. This removal was achieved by using a liquid-vapor separator operated at system pressure (36 bar) prior to the oligomerization reactor. The temperature of the separator was adjusted (373–393 K) such that most of the water would be condensed (97%) with all of the butene remaining in the vapor phase. When decarboxylation of GVL and oligomerization of butene take place over
 and Amberlyst-70, respectively, with a liquid-vapor separator in between, C8+ liquid alkenes can be obtained with 75% overall yield.
and Amberlyst-70, respectively, with a liquid-vapor separator in between, C8+ liquid alkenes can be obtained with 75% overall yield.
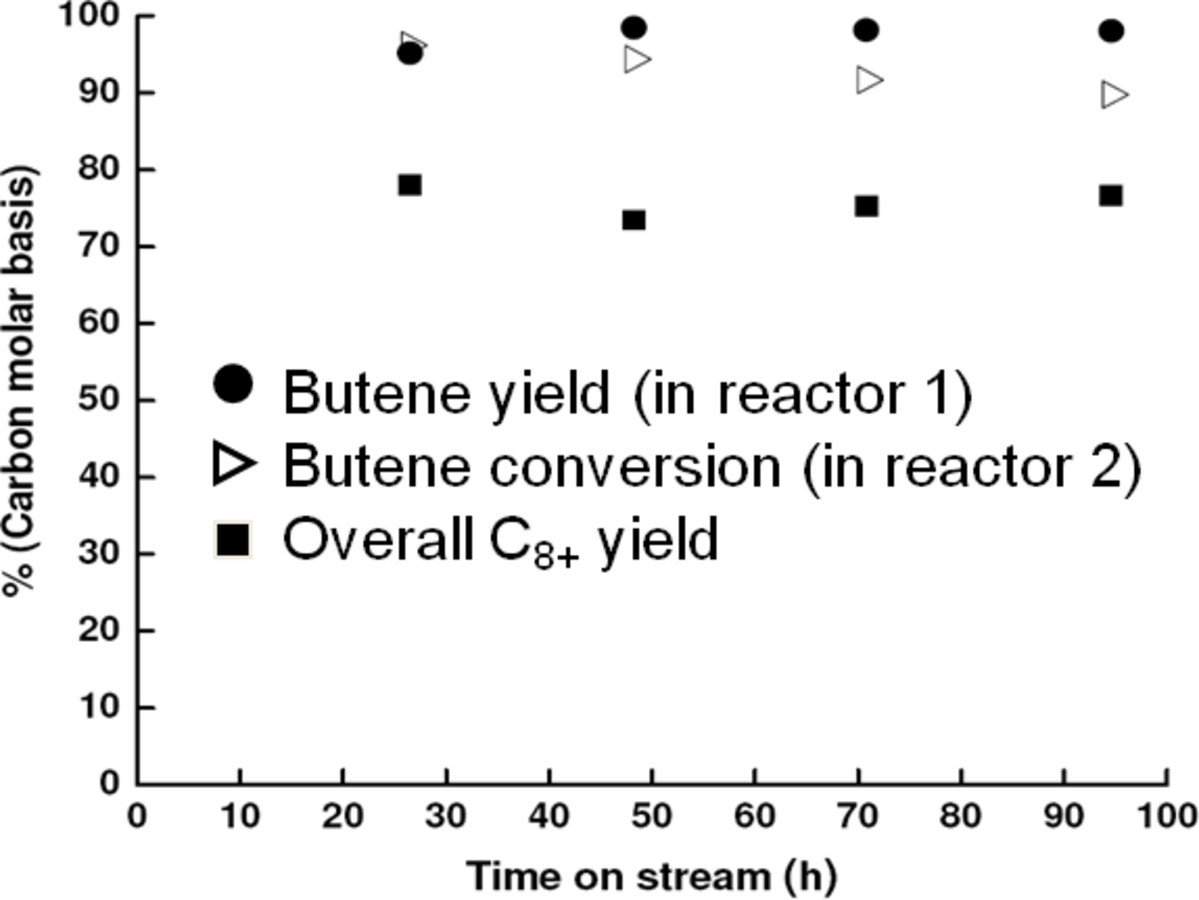
 . First separator is operated at 36 bar and 398 K. Second reactor is at 36 bar, 443 K with Amberlyst-70. Second separator is at 36 bar and 298 K. Adapted from [10]
. First separator is operated at 36 bar and 398 K. Second reactor is at 36 bar, 443 K with Amberlyst-70. Second separator is at 36 bar and 298 K. Adapted from [10]Fig. 10.25: The butene yield from reactor 1, butene conversion in reactor 2 and overall C8+ yields obtained in the operation of the integrated catalytic system for conversion of γ-valerolactone (GVL) to liquid fuels. First reactor is operated at 36 bar, 648 K with
. First separator is operated at 36 bar and 398 K. Second reactor is at 36 bar, 443 K with Amberlyst-70. Second separator is at 36 bar and 298 K. Adapted from [10]
The effluent of the oligomerization reactor is fed to another vapor-liquid separator in series at system pressure and room temperature to obtain alkene oligomers in the condensed phase and
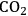 in the gas phase [10]. The process scheme is shown in Figure 10.24 and the butene yield, butene conversion and C8+ yields from the operation of the integrated catalytic system are shown in Figure 10.25. It is important to note that the conversion of GVL to alkene oligomers does not require an external source of hydrogen. Furthermore, the CO2 stream obtained is at high pressure suitable for sequestration. The C8+ oligomers or the corresponding saturated alkanes are ideal to be used in gasoline and jet-fuel applications, while retaining 95% of the energy content of the glucose molecules that make up cellulose.
in the gas phase [10]. The process scheme is shown in Figure 10.24 and the butene yield, butene conversion and C8+ yields from the operation of the integrated catalytic system are shown in Figure 10.25. It is important to note that the conversion of GVL to alkene oligomers does not require an external source of hydrogen. Furthermore, the CO2 stream obtained is at high pressure suitable for sequestration. The C8+ oligomers or the corresponding saturated alkanes are ideal to be used in gasoline and jet-fuel applications, while retaining 95% of the energy content of the glucose molecules that make up cellulose.
10.7 Concluding Remarks
In this chapter, we have outlined strategies for the conversion of carbohydrates to hydrogen or alkane fuels by providing thermodynamics analyses and various examples from recent catalytic processes. First, we indicate how lignocellulosic biomass can be deconstructed to form platform molecules that can be processed further and upgraded to specific fuels and chemicals. Some examples of these platform molecules include glucose, sorbitol and levulinic acid. Even though these species are simpler compared to cellulose, hemicellulose or lignin, they still have sufficient functionality for additional catalytic processing options. These species can be completely deoxygenated to obtain alkanes with 4–6 carbons; however, if alkanes with longer carbon chains than feed molecules are desired, then the deoxygenation reaction should not be carried to completion. Instead, the platform molecules should be processed to produce targeted functional groups (e.g., C=C, C=O, and acid groups), such that these species are easily upgradable through C–C coupling reactions, such as oligomerization, aldol condensation/hydrogenation and ketonization to form higher molecular weight species. We showed with various examples of processes, that this overall strategy can be followed starting with different platform molecules and generating various monofunctional molecules. Therefore, when formulating a new processing strategy, one should decide what useful functional molecules can be obtained, what reactions can be used for controlled removal of oxygen from the platform molecules, and how the C–C coupling between the functional intermediates can be achieved.
Throughout this chapter, we also presented with examples, how the feasibility of a new catalytic process can be explored. As shown for the conversion of carbohydrates to hydrogen, synthesis gas or alkanes, it is important to determine the conditions for which the desired reaction is thermodynamically favorable. In addition, it is required to address the selectivity issues for the desired product by identifying series or parallel competing reactions. Finally, it is necessary to determine overall yields and types of separation steps that would be required.
In order to maximize the yield of the desired products, the factors that control the selectivity should be identified. We demonstrated through examples that selectivity can be altered by changing reaction conditions, the phase of the reaction (liquid- versus gas-phase), types of active sites (metal/acid/base), intrinsic properties of the active sites (e.g., whether C–C or C–O cleavage is favored on a metal), and by using promoters to overcome inhibition and poisoning caused by the reactants, intermediates or products.
We also presented in this chapter the different types of catalytic coupling strategies at different length scales to develop more efficient processing strategies. One easily applicable catalytic coupling strategy is active site coupling that enables the integration of multiple reactions in a single reactor by the use of multifunctional catalysts in a single catalyst bed, and/or by the use of dual catalyst beds. The examples show that there are several factors to be considered to integrate two reactions in a single reactor. First of all, it should be determined if the reaction conditions would allow such an integration. If the integration is possible, then the selectivity in the first reaction should be controlled to provide a suitable feed for the second reaction. Accordingly, the effects of the by-products or co-products of the first reaction on the activity of the second reaction should be investigated. The integration of reactions in such a manner opens new strategies for the efficient production of hydrogen and fuels from lignocellulosic biomass with fewer reaction and separation steps, thereby leading to more cost-competitive biorefining strategies.
Acknowledgments
The results from the University of Wisconsin reported here were supported in part by the U.S. Department of Energy Office of Basic Energy Sciences, by the National Science Foundation Chemical and Transport Systems Division of the Directorate for Engineering, by the DOE Great Lakes Bioenergy Research Center,1 and by the Defense Advanced Research Projects Agency (Surf-cat: Catalysts for Production of JP-8 range molecules from Lignocellulosic Biomass). This chapter was written with the support from Partnerships for International Research and Education (PIRE) program funded by National Science Foundation grant OISE-0730277.
Bibliography
[1] D.A. Simonetti, J.A. Dumesic. Catalytic Strategies for Changing the Energy Content and Achieving C–C Coupling in Biomass-Derived Oxygenated Hydrocarbons ChemSusChem 1: 725-733 (2008): 725-733.
[2] J.N. Chheda, J.A. Dumesic. An Overview of Dehydration, Aldol-Con- densation and Hydrogenation Processes for Production of Liquid Alkanes from Biomass-Derived Carbohydrates Catalysis Today 123: 59-70 (2007): 59-70.
[3] C.-D. Chang, A.J. Silvestri. The Conversion of Methanol and other O-Compounds to Hydrocarbons over Zeolite Catalysts Journal of Catalysis 47: 249-259 (1977): 249-259.
[4] J.N. Chheda, G.W. Huber, G. H.. Liquid-Phase Catalytic Processing of Biomass to Fuels and Chemicals Angewandte Chemie International Edition 46: 7164-7183 (2007): 7164-7183.
[5] T.A. Lloyd, C.E. Wyman. Total Sugar Yields for Pretreatment by Hemicellulose Hydrolysis Coupled with Enzymatic Hydrolysis of the Remaining Solids Bioresource Technology 96: 1967-1977 (2005): 1967-1977.
[6] C.E. Wyman, B.E. Dale, B. D., Elander B.E., E. B., R.T. Elander. Comparative Sugar Recovery Data from Laboratory Scale Application of Leading Pretreatment Technologies to Corn Stover Bioresource Technology 96: 2026-2032 (2005): 2026-2032.
[7] R. Rinaldi, F. Schüth. Acid Hydrolysis of Cellulose as the Entry Point into Biorefinery Schemes ChemSusChem 2(12): 1096-1107 (2009): 1096-1107.
[8] C.E. Wyman, S.R. Decker, S. D., Himmel S.R., H. S., M.E. Himmel. Hydrolysis of Cellulose and Hemicellulose. In: Polysaccharides: Structural Diversity and Functional Versatility New York: Marcel Dekker INC, 2005. .
[9] G.W. Huber, J.N. Chheda, J. C., Barrett J.N.. Production of Liquid Alkanes by Aqueous-Phase Processing of Biomass-Derived Carbohydrates Science 308: 1446-1450 (2005): 1446-1450.
[10] J.Q. Bond, D.M. Alonso, D. A., Wang D.M., W. D.. Integrated Catalytic Conversion of γ-Valerolactone to Liquid Alkenes for Transportation Fuels Science 327: 1110-1114 (2010): 1110-1114.
[11] R.M. West, E.L. Kunkes, E. K., Simonetti E.L.. Catalytic Conversion of Biomass-Derived Carbohydrates to Fuels and Chemicals by Formation and Upgrading of Mono-Functional Hydrocarbon Intermediates Catalysis Today (2008):.
[12] E.I. Gürbüz, E.L. Kunkes, E. K.. Dual-bed Catalyst System for C–C Coupling of Biomass-Derived Oxygenated Hydrocarbons to Fuel-Grade Compounds Green Chemistry 12: 223 (2010): 223.
[13] J. Chaminand, L. Djakovitch, L. D., Gallezot L., G. L., P. Gallezot. Glycerol Hydrogenolysis on Heterogeneous Catalysts Green 6: 359-361 (2004): 359-361.
[14] E.L. Kunkes, D.A. Simonetti, D. S., West D.A., W. D., R.M. West. Catalytic Conversion of Biomass to Monofunctional Hydrocarbons and Targeted Liquid-Fuel Classes Science 322: 417-421 (2008): 417-421.
[15] R.D. Cortright, R.R. Davda, R. D.. Hydrogen from Catalytic Reforming of Biomass-Derived Hydrocarbons in Liquid Water Nature 418: 964 (2002): 964.
[16] M.E. Dry. The Fischer-Tropsch Process: 1950–2000 Catalysis Today 71: 227 (2002): 227.
[17] L. Caldwell. Selectivity in Fischer-Tropsch Synthesis: Review and Recommendations for Further Work. .
[18] D. Tichit, D. Lutic, D. L., Coq D., C. D.. The Aldol Condensation of Acetaldehyde and Heptanal on Hydrotalcite-type Catalysts Journal of Catalysis 219: 167-175 (2003): 167-175.
[19] R.M. West, Z.Y. Liu, Z. L., Peter Z.Y.. Liquid Alkanes with Targeted Molecular Weights from Biomass-Derived Carbohydrates ChemSusChem 1: 417-424 (2008): 417-424.
[20] O. Nagashima, S. Sato, S. S., Takahashi S.. Ketonization of Carboxylic Acids over CO Journal of Molecular Catalysis A 227: 231-239 (2005): 231-239.
[21] J.F. Knifton, J.R. Sanderson, J. S.. Olefin Oligomerization via Zeolite Catalysis Catalysis Letters 28: 223-230 (1994): 223-230.
[22] A.J. Ragauskas, C.K. Williams, C. W., Davison C.K., D. C., B.H. Davison, B. D., Britovsek B.H., B. B., G. Britovsek, G. B., Cairney G., C. G., J. Cairney. The Path Forward for Biofuels and Biomaterials Science 311: 484-489 (2006): 484-489.
[23] P.M. Holmblad, J.H. Larsen, J. L., Chorkendorff J.H., C. J., I. Chorkendorff, I. C., Nielsen I., N. I., L.P. Nielsen. Designing Surface Alloys with Specific Active Sites Catalysis Letters 40: 131-135 (1996): 131-135.
[24] A.P. Wight, M.E. Davis. Design and Preparation of Organic-Inorganic Hybrid Catalysts Chemical Reviews 102: 3589-3614 (2002): 3589-3614.
[25] A. Bhaumik, T. Tatsumi. Organically Modified Titanium-Rich Ti-MCM-41, Efficient Catalysts for Epoxidation Reactions Journal of Catalysis 189: 31-39 (2000): 31-39.
[26] V.L. Budarin, J.H. Clark, J. C., Hardy J.H., H. J., J.J.E. Hardy, J. H.. Starbons: New Starch-Derived Mesoporous Carbonaceous Materials with Tunable Properties Angewandte Chemie International Edition 45: 3782-3786 (2006): 3782-3786.
[27] V.L. Budarin, J.H. Clark, J. C., Luque J.H., L. J., R. Luque. Tunable Mesoporous Materials Optimised for Aqueous Phase Esterifications Green Chemistry 9: 992-995 (2007): 992-995.
[28] V.L. Budarin, J.H. Clark, J. C., Luque J.H.. Versatile Mesoporous Carbonaceous Materials for Acid Catalysis Chemical Communications (2007): 634-636.
[29] V.L. Budarin, R. Luque, R. L., Macquarrie R.. Towards a Bio-Based Industry: Benign Catalytic Esterifications of Succinic Acid in the Presence of Water Chemistry - A European Journal 13: 6914-6919 (2007): 6914-6919.
[30] M.E. Davis. Ordered Porous Materials for Emerging Applications Nature 417: 813-821 (2002): 813-821.
[31] D.J. Macquarrie, D.B. Jackson, D. J., Mdoe D.B.. Organomodified Hexagonal Mesoporous Silicates New Journal of Chemistry 23: 539-544 (1999): 539-544.
[32] D.J. Macquarrie. Direct Preparation of Organically Modified MCM-Type Materials. Preparation and Characterisation of Aminopropyl-MCM and 2-cyanoethyl-MCM Chemical Communications (1996): 1961-1962.
[33] D.J. Macquarrie, D.B. Jackson. Aminopropylated MCMs as Base Catalysts: A Comparison with Aminopropylated Silica Chemical Communications (1997): 1781-1782.
[34] E.L. Margelefsky, R.K. Zeidan, R. Z., Dufaud R.K.. Organized Surface Functional Groups: Cooperative Catalysis via Thiol/Sulfonic Acid Pairing Journal of the American Chemical Society 129: 13691 (2007): 13691.
[35] D. Margolese, J.A. Melero, J. M., Christiansen J.A., C. J.. Direct Syntheses of Ordered SBA-15 Mesoporous Silica Containing Sulfonic Acid Groups Chemistry of Materials 12: 2448-2459 (2000): 2448-2459.
[36] M. Toda, A. Takagaki, A. T., Okamura A., O. A., M. Okamura, M. O.. Green Chemistry: Biodiesel made with Sugar Catalysts Nature 438: 178 (2005): 178.
[37] R.K. Zeidan, V. Dufaud, V. D.. Enhanced Cooperative, Catalytic Behavior of Organic Functional Groups by Immobilization Journal of Catalysis 239: 299-306 (2006): 299-306.
[38] R.K. Zeidan, S.J. Hwang, S. H.. Multifunctional Heterogeneous Catalysts: SBA-15 Containing Primary Amines and Sulfonic Acids Angewandte Chemie International Edition 45: 6332 (2006): 6332.
[39] R.K. Zeidan, M.E. Davis. The Effect of Acid-Base Pairing on Catalysis: An Efficient Acid-Base Functionalized Catalyst for Aldol Condensation Journal of Catalysis 247: 379-382 (2007): 379-382.
[40] R. Zhang, W. Ding, W. D., Tu W.. Mesoporous Silica: An Efficient Nanoreactor for Liquid-Liquid Biphase Reactions Chemistry of Materials 19: 4379-4381 (2007): 4379-4381.
[41] D.Y. Zhao, J.L. Feng, J. F., Huo J.L., H. J., Q.S. Huo, Q. H.. Triblock Copolymer Syntheses of Mesoporous Silica with Periodic 50 to 300 Angstrom Pores Science 279: 548-552 (1998): 548-552.
[42] X.S. Zhao, G.Q. Lu. Modification of MCM-41 by Surface Silylation with Trimethylchlorosilane and Adsorption Study Journal of Physical Chemistry 102: 1556-1561 (1998): 1556-1561.
[43] V. Dufaud, M.E. Davis. Design of Heterogeneous Catalysts via Multiple Active Site Positioning in Organic-Inorganic Hybrid Materials Journal of the American Chemical Society 125: 9403-9413 (2003): 9403-9413.
[44] J.D. Bass, A. Solovyov, A. S., Pascall A.. Acid-Base Bifunctional and Dielectric Outer-Sphere Effects in Heterogeneous Catalysis: A Comparative Investigation of Model Primary Amine Catalysts Journal of the American Chemical Society 128: 3737 (2006): 3737.
[45] J.D. Bass, S.L. Anderson, S. A.. The Effect of Outer-Sphere Acidity on Chemical Reactivity in a Synthetic Heterogeneous Base Catalyst Angewandte Chemie International Edition 42: 5219-5222 (2003): 5219-5222.
[46] J.D. Bass, A. Katz. Bifunctional Surface Imprinting of Silica: Thermolytic Synthesis and Characterization of Discrete Thiol-Amine Functional Group Pairs Chemistry of Materials 18: 1611-1620 (2006): 1611-1620.
[47] I.K. Mbaraka, K.J. McGuire, K. M.. Why are Proteins Charged? Networks of Charge-Charge Interactions in Proteins Measured by Charge Ladders and Capillary Electrophoresis Industrial & Engineering Chemistry Research 45: 3022 (2006): 3022.
[48] I.K. Mbaraka, D.R. Radu, D. R., Lin D.R.. Mesoporous Silicas for the Esterification of Fatty Acids Journal of Catalysis 219: 329-336 (2003): 329-336.
[49] I.K. Mbaraka, B.H. Shanks. Acid Strength Variation due to Spatial Location of Organosulfonic Acid Groups on Mesoporous Silicas Journal of Catalysis 244: 78-85 (2006): 78-85.
[50] I.K. Mbaraka, B.H. Shanks. Conversion of Oils and Fats using Advanced Mesoporous Heterogeneous Catalysts Journal of the American Oil Chemists' Society 83: 79 (2006): 79.
[51] I.K. Mbaraka, B.H. Shanks. Design of Multifunctionalized Mesoporous Silicas for Esterification of Fatty Acids Journal of Catalysis 229: 365-373 (2005): 365-373.
[52] J.M. Notestein, A. Katz. Enhancing Heterogeneous Catalysis through Cooperative Hybrid Organic-Inorganic Interfaces Chemistry - A European Journal 12: 3954-3965 (2006): 3954-3965.
[53] D.L. Klass. Biomass for Renewable Energy, Fuels, and Chemicals. San Diego, CA: Academic Press, 1998
[54] C.J. Barrett, J.N. Chheda, J. C., Huber J.N.. Single-Reactor Process for Sequential Aldol-Condensation and Hydrogenation of Bio- mass-Derived Compounds in Water Applied Catalysis B: Environmental 66: 111-118 (2006): 111-118.
[55] J.I. DiCosimo, G. Torres, G. T.. One-Step MIBK Synthesis: A New Process from 2-Propanol Journal of Catalysis 208: 114-123 (2002): 114-123.
[56] G.W. Huber, R.D. Cortright, R. C.. Renewable Alkanes by Aqueous-Phase Reforming of Biomass-Derived Oxygenates Angewandte Chemie International Edition 43: 1549-1551 (2004): 1549-1551.
[57] A.A. Nikolopoulos, B.W.L. Jang, B. J.. Acetone Condensation and Selective Hydrogenation to MIBK on Pd and Pt Hydrotalcite-Derived Mg-Al Mixed Oxide Catalysts Applied Catalysis A: General 296: 128 (2005): 128.
[58] D.A. Simonetti, J. Rass-Hansen, J. RH., Kunkes J., K. J.. Coupling of Glycerol Processing with Fischer-Tropsch Synthesis for Production of Liquid Fuels Green Chemistry 9: 1073-1083 (2007): 1073-1083.
[59] R.R. Soares, D.A. Simonetti, D. S.. Glycerol as a Source for Fuels and Chemicals by Low-Temperature Catalytic Processing Angewandte Chemie International Edition 45: 3982 (2006): 3982.
[60]
[61] C.H.C. Zhou, J.N. Beltramini, J. B., Fan J.N.. Chemoselective Catalytic Conversion of Glycerol as a Biorenewable Source to Valuable Commodity Chemicals Chemical Society Reviews 37: 527-549 (2008): 527-549.
[62] D.G. Lahr, B.H. Shanks. Effect of Sulfur and Temperature on Ruthe- nium-Catalyzed Glycerol Hydrogenolysis to Glycols Journal of Catalysis 232: 386 (2005): 386.
[63] D.G. Lahr, B.H. Shanks. Kinetic Analysis of the Hydrogenolysis of Lower Polyhydric Alcohols: Glycerol to Glycols Industrial & Engineering Chemistry Research 42: 5467 (2003): 5467.
[64] E.P. Maris, R.J. Davis. Hydrogenolysis of Glycerol over Carbon-Sup- ported Ru and Pt Catalysts Journal of Catalysis 249: 328-337 (2007): 328-337.
[65] E.P. Maris, W.C. Ketchie, W. K., Murayama W.C.. Glycerol Hydrogenolysis on Carbon-Supported PtRu and AuRu Bimetallic Catalysts Journal of Catalysis 251: 281-294 (2007): 281-294.
[66] C. Montassier, J.C. Menezo, J. M., Hoang J.C., H. J.. Aqueous Polyol Conversions on Ruthenium and on Sulfur-Modified Ruthenium Journal of Molecular Catalysis 70: 99 (1991): 99.
[67] C. Montassier, J.C. Menezo, J. M., Moukolo J.C., M. J., J. Moukolo, J. M.. Polyol Conversions into Furanic Derivatives on Bimetallic Catalysts: Cu-Ru, Cu-Pt and Ru-Cu Journal of Molecular Catalysis 70: 65 (1991): 65.
[68] C. Montassier, J.C. Menezo, J. M., Naja J.C., N. J., J. Naja, J. N.. Polyol Conversion into Furanic Derivatives on Bimetallic Catalysts; Nature of the Catalytic Sites Journal of Molecular Catalysis 91: 119-128 (1994): 119-128.
[69] K. Wang, M.C. Hawley, M. H.. Mechanism Study of Sugar and Sugar Alcohol Hydrogenolysis Using 1,3-Diol Model Compounds Industrial & Engineering Chemistry Research 34: 3766 (1995): 3766.
[70] Z. Zhang, Y.-W. Dong, YW. D.. Efficient and Clean Aldol Condensation Catalyzed by Sodium Carbonate in Water Chemistry Letters 32: 966 (2003): 966.
[71] M. Besson, P. Gallezot. Selective Oxidation of Alcohols and Aldehydes on Metal Catalysts Catalysis Today 57: 127-141 (2000): 127-141.
[72] P. Gallezot. Selective Oxidation with Air on Metal Catalysts Catalysis Today 37: 405-418 (1997): 405-418.
[73] J.H.J. Kluytmans, A.P. Markusse, A. M., Kuster A.P., K. A.. Engineering Aspects of the Aqueous Noble Metal Catalysed Alcohol Oxidation Catalysis Today 57: 143-155 (2000): 143-155.
[74] T. Mallat, A. Baiker. Oxidation of Alcohols with Molecular Oxygen on Platinum Metal Catalysts in Aqueous Solutions Catalysis Today 19: 247-284 (1994): 247-284.
[75] D.K. Sohounloue, C. Montassier, C. M.. Catalytic Hydrogenolysis of Sorbitol Reaction Kinetics and Catalysis Letters 22: 391-397 (1983): 391-397.
[76] W.C. Ketchie, M. Murayama, M. M.. Promotional effect of hydroxyl on the aqueous phase oxidation of carbon monoxide and glycerol over supported Au catalysts Topics in Catalysis 44: 307-317 (2007): 307-317.
[77] J.N. Chheda, R. Roman-Leshkov, R. RL.. Production of 5-Hy- droxymethylfurfural and Furfural by Dehydration of Biomass-Derived Mono- and Poly-Saccharides Green Chemistry 9: 342-350 (2007): 342-350.
[78] Y. Roman-Leshkov, C.J. Barrett, C. B., Liu C.J.. Production of Dimethylfuran for Liquid Fuels from Biomass-Derived Carbohydrates Nature 447: 982-985 (2007): 982-985.
[79] Y. Roman-Leshkov, J.N. Chheda, J. C.. Phase Modifiers Promote Efficient Production of Hydroxymethylfurfural from Fructose Science 312: 1933-1937 (2006): 1933-1937.
[80] R.R. Davda, J.W. Shabaker, J. S., Huber J.W., H. J.. A Review of Catalytic Issues and Process Conditions for Renewable Hydrogen and Alkanes by Aqueous-Phase Reforming of Oxygenated Hydrocarbons over Supported Metal Catalysts Applied Catalysis B: Environmental 56: 171-186 (2005): 171-186.
[81] R.R. Davda, J.W. Shabaker, J. S., Huber J.W., H. J.. Aqueous-Phase Reforming of Ethylene Glycol on Silica-Sup- ported Metal Catalysts Applied Catalysis B: Environmental 43: 13-26 (2003): 13-26.
[82] J.H. Sinfelt, D.J.C. Yates. Catalytic Hydrogenolysis of Ethane over the Noble Metals of Group VIII Journal of Catalysis 8: 82 (1967): 82.
[83] D.C. Grenoble, M.M. Estadt, M. E.. The Chemistry and Catalysis of the Water Gas Shift Reaction. 1. The Kinetics over Supported Metal Catalysts Journal of Catalysis 67: 90-102 (1981): 90-102.
[84] M.A. Vannice. The Catalytic Synthesis of Hydrocarbons from Hydrogen/Carbon Monoxide Mixtures over the Group VIII Metals V. The Catalytic Behavior of Silica-Supported Metals Journal of Catalysis 50: 228 (1977): 228.
[85] R. Alcala, M. Mavrikakis, M. M.. DFT Studies for Cleavage of C–C and C–O Bonds in Surface Species Derived from Ethanol on Pt(111) Journal of Catalysis 218: 178-190 (2003): 178-190.
[86] P. Ferrin, D. Simonetti, D. S., Kandoi D., K. D., S. Kandoi, S. K.. Modeling Ethanol Decomposition on Transition Metals: A Combined Application of Scaling and Bronsted-Evans-Polanyi Relations Journal of the American Chemical Society 131: 5809 (2009): 5809.
[87] S. Kandoi, J. Greeley, J. G., Simonetti J., S. J., D. Simonetti. Reaction Kinetics of Ethylene Glycol Reforming over Platinum in the Vapor versus Aqueous Phases Journal of Physical Chemistry C 115: 961 (2011): 961.
[88] R.M. West, D.J. Braden, D. B.. Dehydration of Butanol to Butene over Solid Acid Catalysts in High Water Environments Journal of Catalysis 262: 134-143 (2009): 134-143.
[89] J.M. Encinar, J.F. Gonzales, J. G.. Biodiesel from Used Frying Oil. Variables Affecting the Yields and Characteristics of the Biodiesel Industrial & Engineering Chemistry Research 44: 5491-5499 (2005): 5491-5499.
[90] E. Christoffersen, P. Liu, P. L., Ruban P., R. P.. Anode Materials for Low Temperature Fuel Cells Journal of Catalysis 199: 123-131 (2001): 123-131.
[91] E. Iglesia, S.C. Reyes, S. R., Madon S.C.. Selectivity Control and Catalyst Design in the Fischer-Tropsch Synthesis: Sites, Pellets and Reactors Advances in Catalysis 39: 221-302 (1993): 221-302.
[92] E.L. Kunkes, D.A. Simonetti, D. S., Dumesic D.A., D. D., J.A. Dumesic, J. D.. The Role of Rhenium in the Conversion of Glycerol to Synthesis Gas over Carbon Supported Platinum-Rhenium Catalysts Journal of Catalysis 260: 164-177 (2008): 164-177.
[93] V. Pallassana, M. Neurock. Reaction Paths in the Hydrogenolysis of Acetic Acid to Ethanol over Pd(111), Re(0001), and PdRe Alloys Journal of Catalysis 209: 289-305 (2002): 289-305.
[94]
[95] C.H. Bartholomew, R.J. Farrauto. Fundamentals of Industrial Catalytic Processes. Hoboken, NJ: Wiley, 2006
[96]
[97]
[98] A.G. Gayubo, A.T. Aguayo, A. A., Atutxa A.T., A. A.. Transformation of Oxygenate Components of Biomass Pyrolysis Oil on a HZSM-5 Zeolite. I. Alcohols and Phenols Industrial & Engineering Chemistry Research 43: 2610-2618 (2004): 2610-2618.
[99] J. Lopez, J. Valente, J. V., Sanchez J., S. J.. Hydrogen Transfer Reduction of 4-Tert-Butylcyclohexanone on Basic Sol- ids Journal of Catalysis 208: 30-37 (2002): 30-37.
[100] S.G. Powell, A.T. Nielsen. Condensation of Butanal with 4-Heptanone and 3-Hexanone and Attempted Condensation of 2-Ethyl-2-hexenal with 4-Heptanone Journal of the American Chemical Society 70: 3627 (1948): 3627.
[101] E.L. Kunkes, E.I. Gürbüz, E. G.. Vapour-Phase C–C Coupling Reactions of Biomass-Derived Oxygenates over Pd/CeZrOx Catalysts Journal of Catalysis 206: 236-249 (2009): 236-249.
[102] J.I. DiCosimo, V.K. Diez, V. D., Xu V.K., X. V.. Structure and Surface and Catalytic Properties of Mg-Al Basic Oxides Journal of Catalysis 178: 499-510 (1998): 499-510.
[103] V.K. Diez, J.I.D. Cosimo, J. C.. Study of the Citral/Acetone Reaction on Mg Applied Catalysis A: General 345: 143-151 (2008): 143-151.
[104] E.I. Gürbüz, E.L. Kunkes, E. K.. Integration of C–C Coupling Reactions of Biomass-Derived Oxygenates to Fuel-Grade Compounds Applied Catalysis B: Environmental 94: 134-141 (2010): 134-141.
[105] J.J. Bozell, L. Moens, L. M., Elliott L., E. L., D.C. Elliott, D. E., Wang D.C.. Production of Levulinic Acid and Use as a Platform Chemical for Derived Products Resources, Conservation and Recycling 28: 227-239 (2000): 227-239.
[106]
[107]
[108] J.C. Serrano-Ruiz, D.J. Braden, D. B., West D.J.. Conversion of Cellulose to Hydrocarbon Fuels by Progressive Removal of Oxygen Applied Catalysis B: Environmental 100: 184-189 (2010): 184-189.
[109] M. Mascal, E.B. Nikitin. High-Yield Conversion of Plant Biomass into the Key Value-Added Feedstocks 5-(Hydroxymethyl)Furfural, Levulinic Acid, and Levulinic Esters via 5-(Chloromethyl)Furfural Green Chemistry 12: 370-373 (2010): 370-373.
[110] P. Wang, S. Zhan, S. Z.. Production of Levulinic Acid from Cellulose Catalyzed by Environmental-Friendly Catalysts Advanced Materials Research 96: 183 (2010): 183.
[111] F. Tao, H. Song, H. S.. Catalytic Conversion of Cellulose to Chemicals in Ionic Liquid Carbohydrate Research 346: 58-63 (2011): 58-63.
[112] J. Horvat, B. Klaic, B. K., Metelko B.. Mechanism of Levulinic Acid Formation Tetrahedron Letters 2: 2111-2114 (1985): 2111-2114.
[113] J.P. Lange, R. Price, R. P., Ayoub R., A. R., P.M. Ayoub, P. A.. Valeric Biofuels: A Platform of Cellulosic Transportation Fuels Angewandte Chemie International Edition 49: 4479-83 (2010): 4479-83.
[114] J.C. Serrano-Ruiz, D. Wang, D. W.. Catalytic Upgrading of Levulinic Acid to 5-Nonanone Green Chemistry 12: 574-577 (2010): 574-577.
[115] H. Heeres, R. Handana, R. H., Chunai R., C. R., D. Chunai. Combined Dehydration/(Transfer)-Hydrogenation of C6-Sugars (D-Glucose and D-Fructose) to γ-Valerolactone using Ruthenium Catalysts Green Chemistry 11: 1247-1255 (2009): 1247-1255.
[116] L.E. Manzer. Catalytic Synthesis of α-Methylene-γ-Valerolactone: A Bio- mass-Derived Acrylic Monomer Applied Catalysis A: General 272: 249-256 (2004): 249-256.
[117] J.P. Lange, J.Z. Vestering, J. V.. Towards “Bio-Based” Nylon: Conversion of γ-Valerolactone to Methyl Pentenoate under Catalytic Distillation Conditions Chemical Communications 33: 3488-3490 (2007): 3488-3490.
[118] I.T. Horvath, H. Mehdi, H. M., Fabos H., F. H.. γ-Valerolactone - A Sustainable Liquid for Energy and Carbon-Based Chemicals Green Chemistry 10: 238-242 (2008): 238-242.
[119] D.M. Alonso, J.Q. Bond, J. B., Serrano-Ruiz J.Q.. Production of Liquid Hydrocarbon Transportation Fuels by Oligomerization of Biomass-Derived C(9) Alkenes Green Chemistry 12: 992-999 (2010): 992-999.
[120] J.Q. Bond, D.M. Alonso, D. A., West D.M.. γ-Valerolactone Ring-Opening and Decarboxylation over SiO Langmuir 26: 16291-16298 (2010): 16291-16298.
[121] J.Q. Bond, D. Wang, D. W., Alonso D.. Interconversion between γ-Valerolactone and Pentenoic Acid Combined with Decarboxylation to form Butene over Silica/Alumina Journal of Catalysis 281: 290-299 (2011): 290-299.