Chapter structure
- 11.1 Introduction
- 11.2 Adsorption of Carbohydrates and Their Derivatives on
Solid Surfaces - 11.3 Selective Bond Cleavage
- 11.4 Aqueous Phase Considerations
- 11.5 Novel Catalytic Materials for Biomass Conversion
- 11.6 Hydrothermally Stable Catalytic Materials
- 11.7 Impurity Tolerant Catalysts
- 11.8 Novel Reaction Systems
- 11.9 Summary
- Bibliography
11.1 Introduction
Designing heterogeneous catalysts for the conversion of biorenewable feedstocks, e.g., carbohydrates, lipids, and lignins, creates new challenges and opportunities for catalyst technology. The development of the science and technology of heterogeneous catalysis has been performed largely in concert with the development of the science and technology associated with the production of fuels and petrochemicals from crude oil and natural gas with the one notable exception being the treatment of the gaseous effluent from combustion processes. In general, the crude oil and natural gas-based feedstocks used for fuel and petrochemical production are not particularly reactive, so they typically are “activated.” The most common means of activation is through cracking-type reactions. This cracking is performed at elevated temperatures and leads to shorter chain molecules and compounds that contain a small fraction of unsaturated C–C bonds, which can be exploited for subsequent catalytic transformations. In contrast, biorenewable feedstocks already contain quite a number of functional groups that can be used for chemical transformations, which necessitates a new paradigm of how to design heterogeneous catalysts that can do selective chemistry in the presence of multiple reactive functional groups within a reactant molecule.
The fine and specialty chemical market has been confronted with the challenge of selectively converting substrates containing multiple reactive groups, which has commonly been addressed through the use of homogeneous catalysts or biocatalysts. Given the high value of fine and specialty chemicals, these types of catalyst systems can be economically deployed. However, the cost, temperature limitations, and separation issues that can be created by using homogeneous catalysts and biocatalysts will undoubtedly restrict their use in biorenewable feedstock conversions when the target products are lower-valued products such as fuel and industrial chemicals.
While the reactive moiety generated in cracking hydrocarbons for use in subsequent reactions is an unsaturated C–C bond, the multiple reactive functional groups within biorenewable feedstocks usually involve oxygen. Therefore, chemical transformations of biorenewable feedstocks will need to be performed in the presence of hydroxyl, carbonyl, carboxylic acid, ester, and/or ether groups. One outcome from this difference is that well established heterogeneous catalyst technology developed for hydrocarbon feedstocks, which are efficient for selective hydrogenation, selective oxidation, isomerization, reforming, etc., has not been designed for the dehydration, decarboxylation, decarbonylation, hydrogenolysis, esterification, ketonization, etc., reactions that are needed for converting biorenewable feedstocks to desirable products.
A second challenge introduced due to the presence of multiple functional groups in biomass-derived compounds is the poor volatility of the compounds at relevant reaction temperatures. This property commonly leads to the requirement of condensed phase reaction systems, which is in marked contrast to the primarily gas-phase reaction systems used for hydrocarbon processing. The resulting liquid-solid interfaces place new demands on catalytic materials relative to catalytic properties, catalyst stability and transport properties, which are further exacerbated when the condensed phase solvent is water. Due to their high oxygen content, biorenewable feedstocks are effectively hydrogen deficient relative to hydrocarbons, so three-phase reaction systems consisting of a solid catalyst, liquid solvent/reactant mixture and hydrogen gas will commonly need to be employed to generate products that replace those from hydrocarbons.
As discussed in previous chapters, biorenewable feedstocks can take many different forms, so it is important to define the range of molecules to be discussed in the current chapter. The primary focus of the heterogeneous catalyst design issues to be considered will be carbohydrates and their derivatives as these represent the most significant departure in chemistry from hydrocarbon feedstocks. There will be limited discussion of triglyceride and fatty acid reactions as examples for demonstrating some of the concepts. Increasingly, lignin and its aromatic building blocks are receiving attention, but they will not be discussed explicitly in this chapter.
Carbohydrates exist in nature primarily as polymeric species, so these polymers must be sufficiently deconstructed to create soluble compounds if heterogeneous catalysts are used for further transformations. The deconstruction of carbohydrates can be loosely categorized by the temperature in which it is performed (see Figure 11.1). Under high temperature conditions (~800 °C), carbohydrates can be gasified to produce CO and
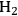 also known as syngas. The catalytic chemistry associated with converting syngas to fuels and chemicals has received significant attention over quite a number of years. Neglecting tar compounds, trace contaminants and the CO:H2 ratio (admittedly, these are important properties that in practice cannot be ignored), syngas is a carbon-based feedstock agnostic, so obtaining it from carbohydrate gasification does not introduce new syngas chemistry. At intermediate temperatures (350–550 °C), solid carbohydrates can be converted to a liquefied form. High-pressure liquefaction can be accomplished at about 350 °C and pyrolysis, either fast thermal or catalytic, typically employs temperatures ranging from 450–550 °C. These intermediate temperature processes when using established technology cause non-selective deconstruction of carbohydrate polymers leading to a liquid product, known as bio-oil, that is composed of numerous oxygenated chemical species. Given the compositional complexity of bio-oil, attention has largely been focused on upgrading it to fuels rather than for converting and purifying it to specific chemical products. Selective deconstruction of carbohydrates can be achieved at 35–170 °C by hydrolyzing the glycosidic bond between glucosyl rings. This approach, which can utilize enzymes, ionic liquids, or acids, results in the liberation of predominantly monomeric carbohydrates if the proper reaction conditions are imposed. The sugars released, glucose, xylose, arabinose, and mannose, can in principle be converted to fuels or chemicals. The primary focus of the discussion in the current chapter will be on the catalyst properties that need to be considered in the conversion of the monomeric sugars from hydrolysis or the oxygenated species in bio-oil into fuels and chemicals.
also known as syngas. The catalytic chemistry associated with converting syngas to fuels and chemicals has received significant attention over quite a number of years. Neglecting tar compounds, trace contaminants and the CO:H2 ratio (admittedly, these are important properties that in practice cannot be ignored), syngas is a carbon-based feedstock agnostic, so obtaining it from carbohydrate gasification does not introduce new syngas chemistry. At intermediate temperatures (350–550 °C), solid carbohydrates can be converted to a liquefied form. High-pressure liquefaction can be accomplished at about 350 °C and pyrolysis, either fast thermal or catalytic, typically employs temperatures ranging from 450–550 °C. These intermediate temperature processes when using established technology cause non-selective deconstruction of carbohydrate polymers leading to a liquid product, known as bio-oil, that is composed of numerous oxygenated chemical species. Given the compositional complexity of bio-oil, attention has largely been focused on upgrading it to fuels rather than for converting and purifying it to specific chemical products. Selective deconstruction of carbohydrates can be achieved at 35–170 °C by hydrolyzing the glycosidic bond between glucosyl rings. This approach, which can utilize enzymes, ionic liquids, or acids, results in the liberation of predominantly monomeric carbohydrates if the proper reaction conditions are imposed. The sugars released, glucose, xylose, arabinose, and mannose, can in principle be converted to fuels or chemicals. The primary focus of the discussion in the current chapter will be on the catalyst properties that need to be considered in the conversion of the monomeric sugars from hydrolysis or the oxygenated species in bio-oil into fuels and chemicals.
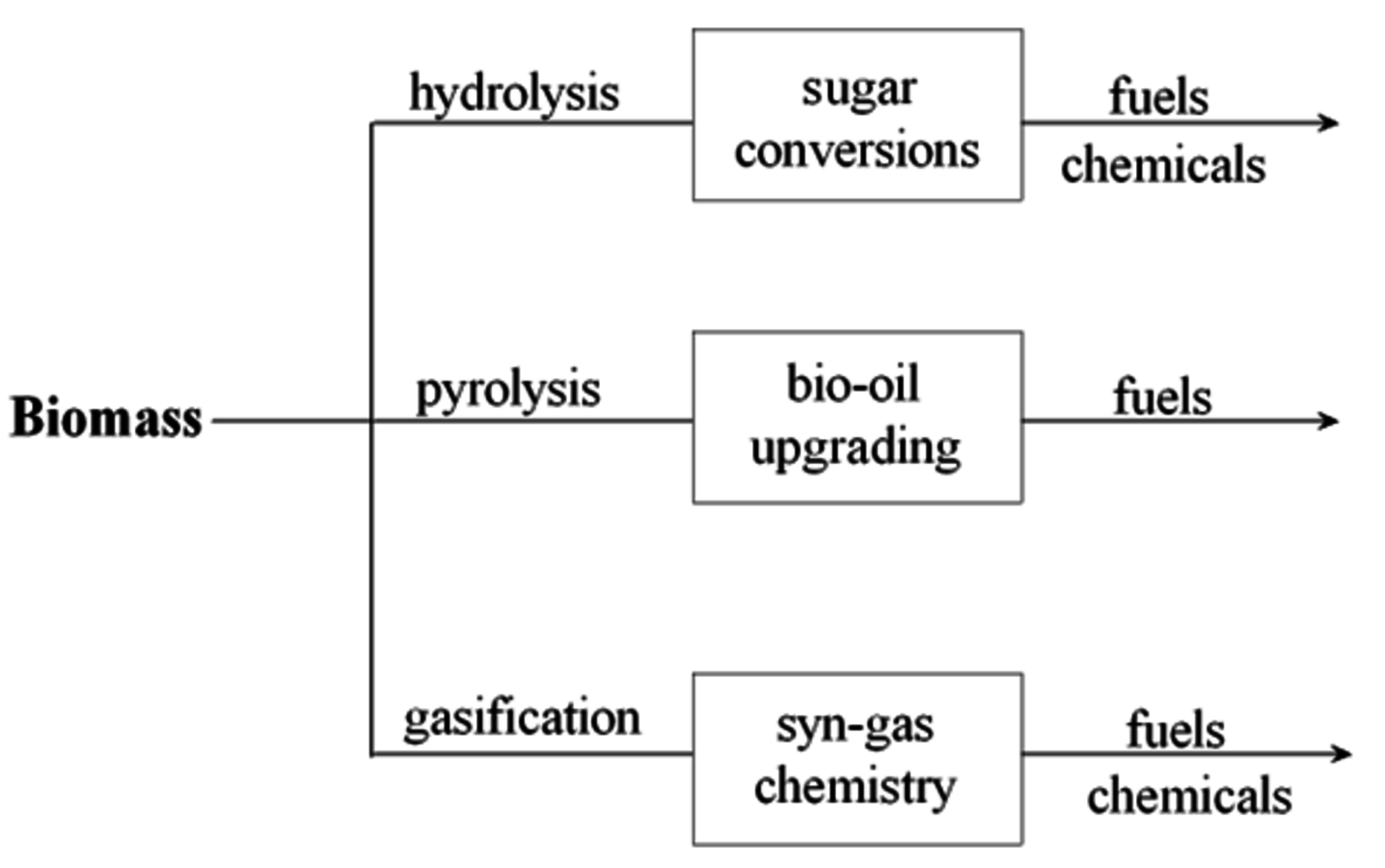
Fig. 11.1: Biomass deconstruction and upgrading pathways
The conversion of carbohydrate-derived species in the condensed phase using heterogeneous catalysts introduces a broad array of interesting questions ranging from those concerning fundamental reaction mechanisms to reaction systems. How do molecules with a high level of oxygen functionality adsorb on metal/metal oxide surfaces? Can specific C–C or C–O bonds be targeted for conversion when multiple reactive functional groups are present? How will water play a role in these condensed phase reactions? How should catalytic materials be modified for use in converting carbohydrates and their derivatives? Can solid catalysts be constructed that can withstand hydrothermal conditions? Can heterogeneous catalyst systems be designed to deal with biorenewable feedstocks impurities? Can the catalytic reaction systems be coupled with separations to improve efficiencies? Rather than categorizing by specific reaction classes, this chapter will discuss these questions. Examples of some studies that have begun to examine these questions will be given as illustrations.
11.2 Adsorption of Carbohydrates and Their Derivatives on
Solid Surfaces
Through a combination of experimental and theoretical work, the adsorption of hydrocarbons on catalytic surfaces has received sufficiently extensive attention such that the interaction between unsaturated C–C bonds and metal or metal oxide surfaces is reasonably understood. The interaction of other functional groups such as hydroxyls with catalytic surfaces has also been examined. However, carbohydrates and their derivatives contain multiple functional groups and the adsorption behavior of these types of molecules has only received a limited amount of attention. While only considering the adsorption interaction of a single molecule possessing multiple functional groups with a metal or metal oxide surface is challenging to characterize, the added complexity of probing this adsorption behavior under reaction conditions is even more daunting. Excellent in situ characterization techniques have been developed that can probe adsorbate-surface interactions under gas-phase conditions, but effective in situ characterization approaches for characterizing condensed phase reaction systems are only beginning to be developed.
Rather than attempting an exhaustive treatment of all types of functional groups that can arise in carbohydrates and species derived from carbohydrates, the interaction between hydroxyl groups and surfaces will be discussed for the case when multiple hydroxyl groups are present in the adsorbing molecule. A limiting case of this situation is the adsorption of 1,2-propylene glycol and glycerol. Peereboom et al. [1] examined the adsorption of these species on Ru at ambient temperature due to its implication on the hydrogenolysis of glycerol to produce propylene glycol. Over extended adsorption times, they found that the total amount of glycerol or propylene glycol adsorbed on a clean Ru surface was roughly equivalent. The competitive adsorption of the two species was also evaluated. First, the clean Ru surface was exposed to an aqueous solution of one of the species and then the second was added at a later time point (Figure 11.2). For the data in Figure 11.2A), Ru was exposed to a propylene glycol aqueous solution for about 17 hours and then glycerol of equal concentration was introduced. An uptake of glycerol was immediately observed. In contrast, the initial addition of a glycerol aqueous solution followed by propylene glycol introduction at about 17 hours, as shown in Figure 11.2B), was found to inhibit the adsorption of propylene glycol. This apparent stronger adsorption affinity for glycerol was consistent with previous literature. Based on fitting kinetic parameters to reaction data for glycerol hydrogenolysis using a Ru supported on a carbon catalyst, the glycerol adsorption constant was found to be five times higher than the constant for propylene glycol [2]. While purely speculative, the significant difference in adsorption behavior would seem to be related to either the presence of three hydroxyl groups in glycerol or the alkyl group at the end of propylene glycol.
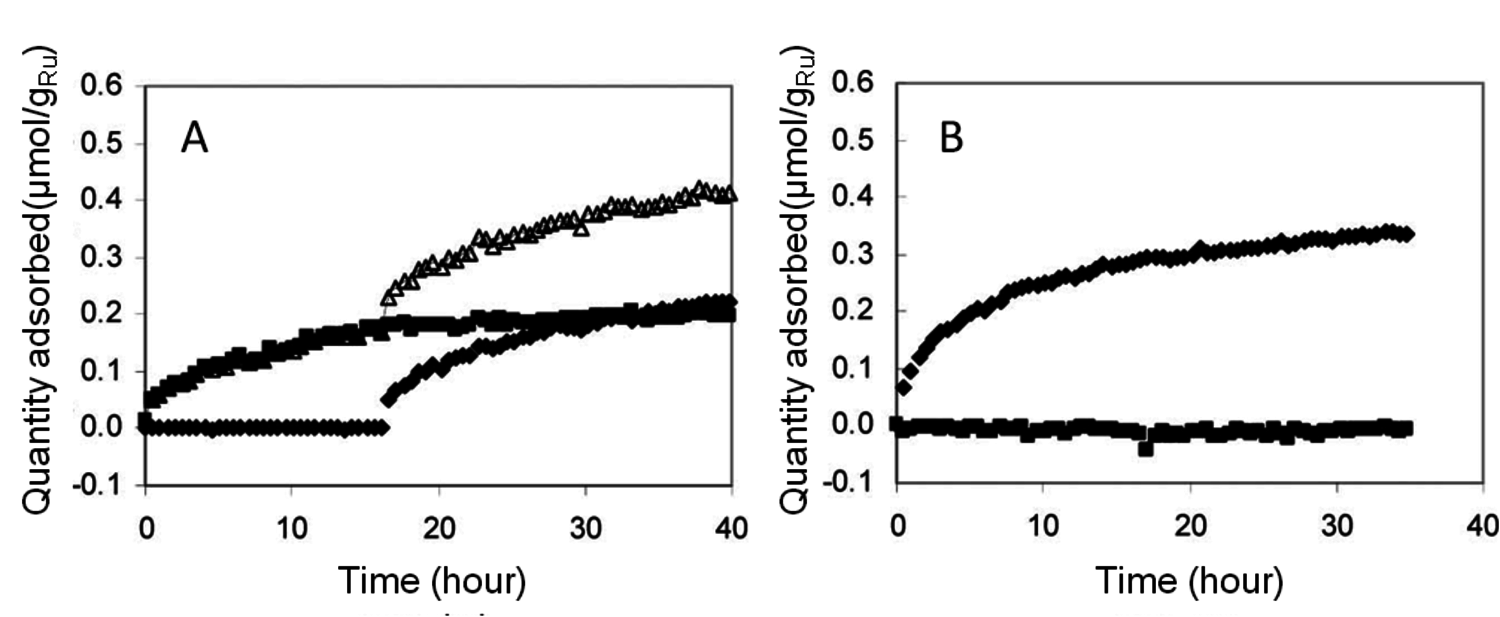
Fig. 11.2: Polyol adsorption (propylene glycol - squares; glycerol - diamonds; total moles adsorbed - triangles) on Ru at ambient temperature: (A) initial propylene glycol adsorption followed by glycerol introduction at 15 hours, (B) initial glycerol adsorption followed by propylene glycol introduction at 15 hours [1]
As demonstrated by the above example, the adsorption behavior for relatively simple polyhydroxylated species is not well understood. When considering the adsorption behavior of carbohydrates, additional complexity is introduced by the fact that carbohydrates can exist in different conformations. In the case of D-glucose, it exists in aqueous solution at ambient temperature almost exclusively in the glucopyranose form in which the five carbons and an oxygen form a six-membered ring with the oxygen linkage between the C1 and C5 carbons. However, when D-glucose is allowed to equilibrate in an aqueous solution, α- and β-glucopyranose isomers exist in the ratio of 36:64, where the isomers correspond to whether the hydroxyl group on the C1 carbon is above or below the plane of the ring. In contrast, fructose in an aqueous solution exists at ambient temperature in four cyclic and one acyclic isomer form, with six-membered ring fructopyranose (70%) and five-membered ring fructofuranose (22%) being the dominant forms. Although it might be expected that the interaction between a specific carbohydrate, such as D-glucose, and a catalytic surface would lead to a single type of adsorbed species, which presumably would be the most favorable energetically, no systematic study has been performed to validate this presumption.
Given the stereogenic centers present in carbohydrates, a family of carbohydrate stereoiosmers exist that have the same molecular formula. This family represents molecules with different configurations. Since carbohydrates with different configurations do not spontaneously isomerize to a different configuration, the different stereoisomers can be used to gain some information about how carbohydrates adsorb.
One such example is a study that examined the adsorption behavior of a range of sugars in aqueous solution onto hydrous zirconium oxide and iron oxide at ambient temperature [3]. In an experimental approach that employed Langmuir monolayer adsorption, aldohexoses, aldopentoses, and ketohexoses as well as several of their derivatives were evaluated (shown in Figure 11.3 are the sugars used in the study). The Langmuir equilibrium constant as well as the maximum amount of a sugar required to form a monolayer on the solid surface were found to be strongly dependent on the stereoisomer used. D-ribose had a Langmuir equilibrium constant that was five times that for D-glucose and a maximum amount of molecules in the resulting monolayer that was 50% higher. A general trend was found in which the stereochemistry of the three neighboring hydroxyl groups on either the C2, C3, C4 carbon atoms for aldohexoses or the hydroxyl groups on the C2, C3, C4 carbon atoms in ketohexoses strongly correlated with the resulting adsorption behavior. Interestingly, the differences in adsorption behavior between the sugars examined was significantly more pronounced when the surface was hydrous zirconium oxide rather than hydrous iron oxide. The surface-specific response was speculated to be due to the difference between the ionic radii of the metal ions in the two hydrous metal oxides.
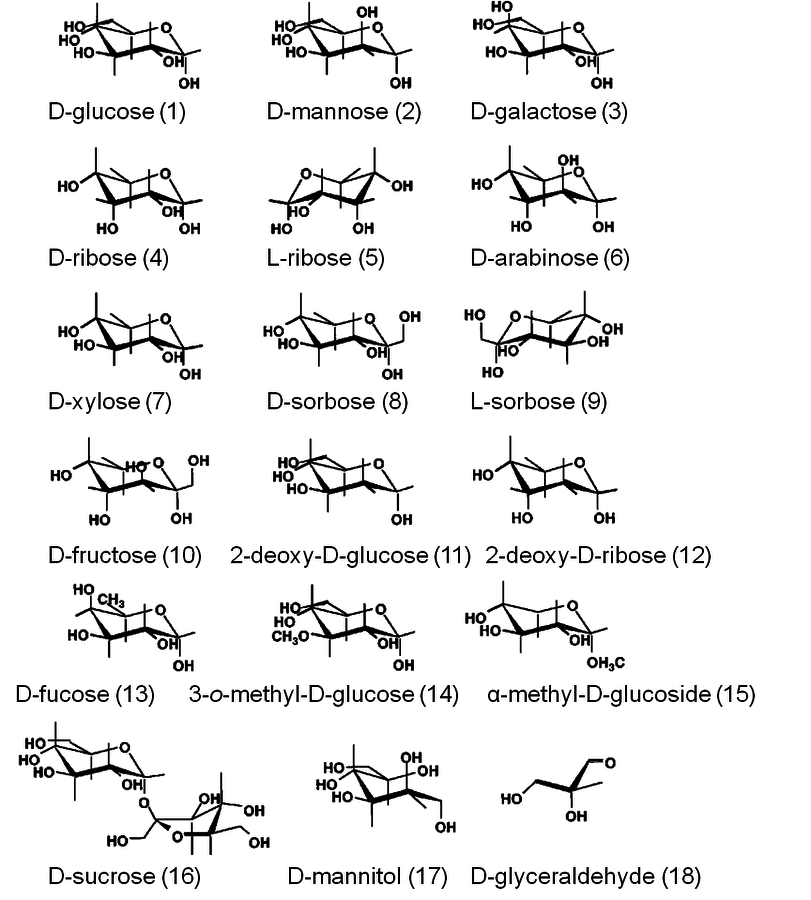
Fig. 11.3: Carbohydrate species used in a Langmuir adsorption study over hydrous zirconium oxide and iron oxide [3]
As the preferred conformation of an adsorbed carbohydrate could be a function of temperature, ambient temperature adsorption studies of carbohydrates might not directly correlate with their adsorbed conformation at the elevated temperatures used in reactions. Unfortunately, no reliable methods are currently available to examine carbohydrate adsorption from an aqueous solution at elevated temperatures. An indirect method to examine adsorption behavior is through reactivity studies.
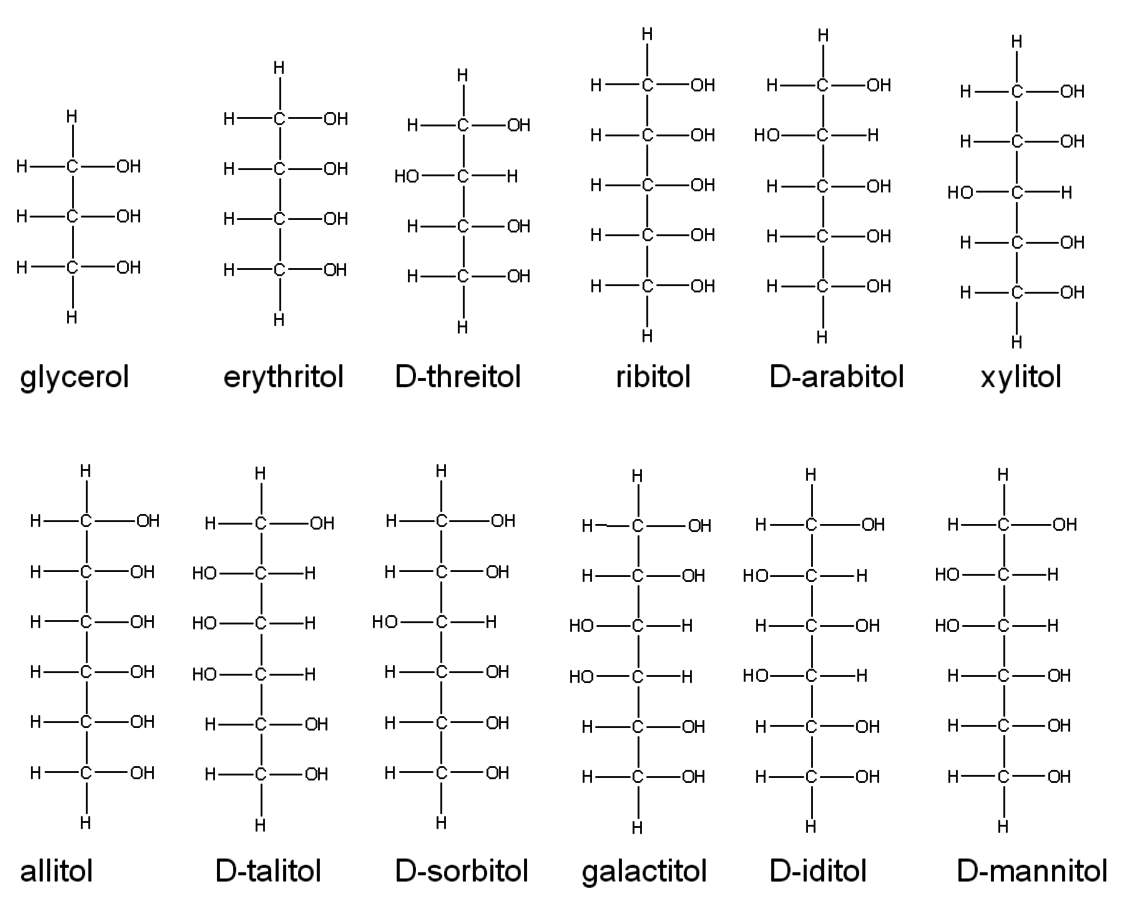
Fig. 11.4: Fischer projections of the alditols from C3 to C6
An example of this approach was a study of the initial reactivity of alditol stereoisomers, corresponding to the hydrogenated forms of sugars, under hydrogenolysis conditions [4]. In this study, the relative initial rate of disappearance of alditols in an aqueous system was compared over a Ru on a carbon catalyst. Shown in Figure 11.4 are the Fischer Projections of the C3–C6 alditols. As several of the C6 alditols have limited solubility in water, Table 11.1 gives the initial hydrogenolysis rates for those with sufficient solubility grouped according to their carbon number. As can be seen from this grouping, the intital reaction rates did not correlate with the alditol carbon number as stereoisomers had different reactivities. As demonstrated in the carbohydrate adsorption study discussed above, the stereoisomers for a given carbon number likely have somewhat different adsorption characteristics, which would be expected to be manifest in their resulting reactivity.
| Compound | Reaction Rate (205 °C, mmol/min) |
Reaction Rate (240 °C, mmol/min) |
|---|---|---|
| Glycerol | 3.9 | 10.3 |
| Erythritol | 3.4 | 10.1 |
| Threitol | 2.0 | 5.0 |
| Ribitol | 3.3 | 11.0 |
| Arabitol | 3.0 | 9.5 |
| Xylitol | 2.5 | 6.4 |
| Mannitol | 2.8 | 7.8 |
| Sorbitol | 2.5 | 6.7 |
| Galactitol | N/A | 3.5 |
Tab. 11.1: Initial reaction rates for alditols over a Ru/C catalyst (0.5 M alditol concentration, 100 bar H2, and pH adjusted to 11.5 using CaO)
Tab. 11.1: Initial reaction rates for alditols over a Ru/C catalyst (0.5 M alditol concentration, 100 bar H2, and pH adjusted to 11.5 using CaO)
One could ask whether the reactivity data could correlate better to configuration rather than carbon number. Consider the three pentitols stereoisomers as shown again in their Fischer Projections in Figure 11.5.
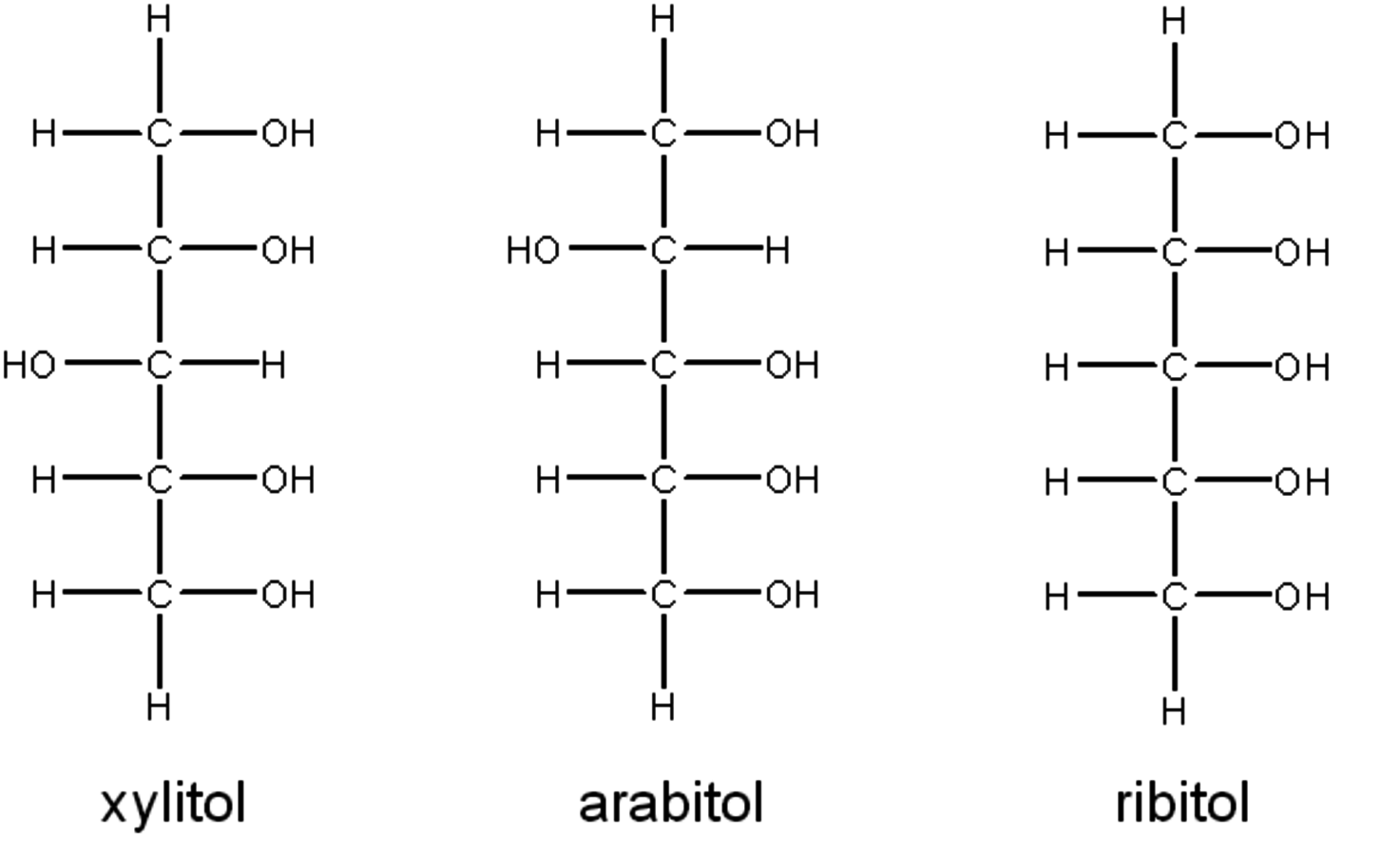
Fig. 11.5: Fischer projections of the C5 alditols, which provide a comparison of their different configurations
These molecules consist of three chiral carbons as the two terminal carbons can freely rotate. A simple classification would be to count the number of hydroxyl group inversions for the chiral carbons, corresponding to C2, C3 and C4. For ribitol, all of the hydroxyl groups reside on the same side, so it can be categorized as having zero inversions. Using the same categorization approach, xylitol possesses two inversions and arabitol has one. Shown in Table 11.2 are the initial reaction rate data now categorized by the number of inversions rather than carbon number (glycerol has no chiral carbon, so it cannot have an inversion). While not perfectly correlated, this categorization does a better job of lumping similar observed reaction rates than does carbon length.
| Alditol | Reaction Rate (mmol/min) |
Inversion | Alditol Length |
|---|---|---|---|
| Glycerol | 10.3 | 0 | 3 |
| Erythritol | 10.2 | 0 | 4 |
| Ribitol | 11.0 | 0 | 5 |
| Threitol | 5.0 | 1 | 4 |
| Arabitol | 9.5 | 1 | 5 |
| Mannitol | 7.8 | 1 | 6 |
| Xylitol | 6.4 | 2 | 5 |
| Sorbitol | 6.7 | 2 | 6 |
| Galactitol | 3.5 | 2 | 6 |
Tab. 11.2: Groupings of initial alditol reaction rates relative to their configurations (Ru/C, 0.5 M polyol concentration, 100 bar H2, and pH adjusted to 11.5 using CaO)
Tab. 11.2: Groupings of initial alditol reaction rates relative to their configurations (Ru/C, 0.5 M polyol concentration, 100 bar H2, and pH adjusted to 11.5 using CaO)
A second aspect that could play a role in the adsorption and reactivity of the alditols is the conformation of the molecules upon adsorption. C5 and C6 alditols can adopt either zig-zag or sickle conformation in solution. Shown in Figure 11.6 are xylitol and ribitol in these two conformations. As can be seen from the figure, xylitol in the zig-zag conformation has all of the hydroxyl groups on the chiral carbons pointing in the same direction, while in the sickle conformation, at most two hydroxyl groups on the chiral carbons are in the same direction. This situation is exactly reversed for ribitol. Under ambient temperatures and in aqueous solution, NMR analysis has shown that the zig-zag conformation dominates for alditols [5]. When the complexation of xylitol and ribitol with Ca(II) was examined at ambient temperature, xylitol was found to more readily complex than ribitol. This difference might be attributed to the hydroxyl group location for xylitol, which point in the same direction in the zig-zag conformation leading to more potential interaction points between the molecule and the surface. If it is presumed that the reactivity of xylitol and ribitol is correlated with adorption, then the higher reactivity of ribitol might suggest that the preferred adsorption conformation at the elevated temperatures used for hydrogenolysis is the sickle form as ribitol has all of the hydroxyls on the chiral carbons pointing in the same direction. Clearly, this postulate is highly speculative, but it points to the lack of understanding that currently exists with regard to the adsorption behavior of carbohydrates and their derivatives on solid surfaces under reaction conditions.
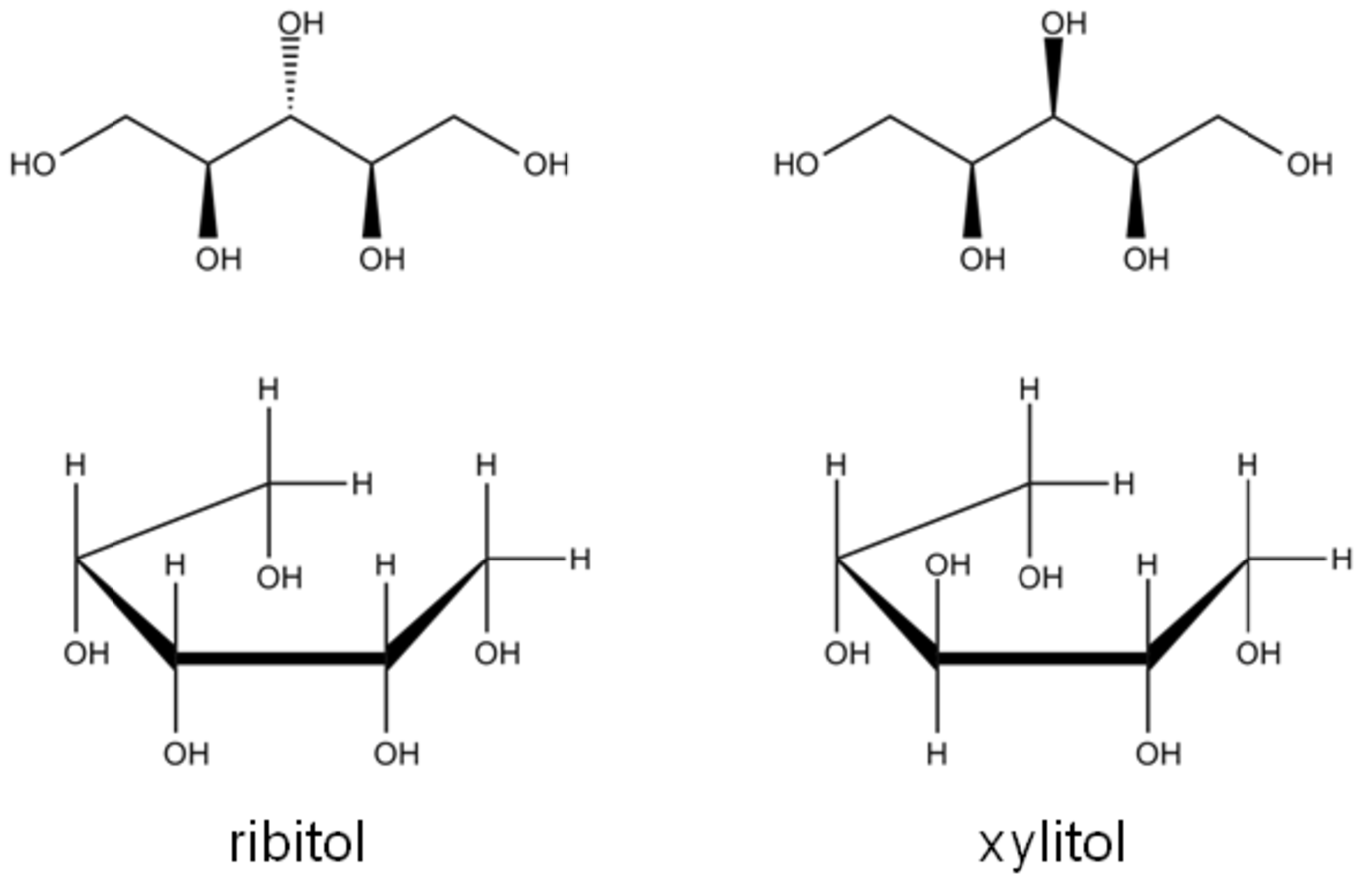
Fig. 11.6: Possible conformations for ribitol and xylitol, which are zig-zag (top) and sickle (bottom)
11.3 Selective Bond Cleavage
For most targeted products from biomass, the biorenewable feedstocks will typically contain excess oxygen, so there is a desire to develop catalytic transformations that can selectively remove oxygen. Examples of reaction classes that have been examined for oxygen removal from biorenewable feedstocks are dehydration, decarbonylation/decarboxylation, condensation and ketonization. Not only are these reactions not the typical ones that have been the focus of catalyst development for hydrocarbon processing, but they have the added complication in biorenewable feedstock conversion of needing to be achieved in the presence of multiple reactive groups.
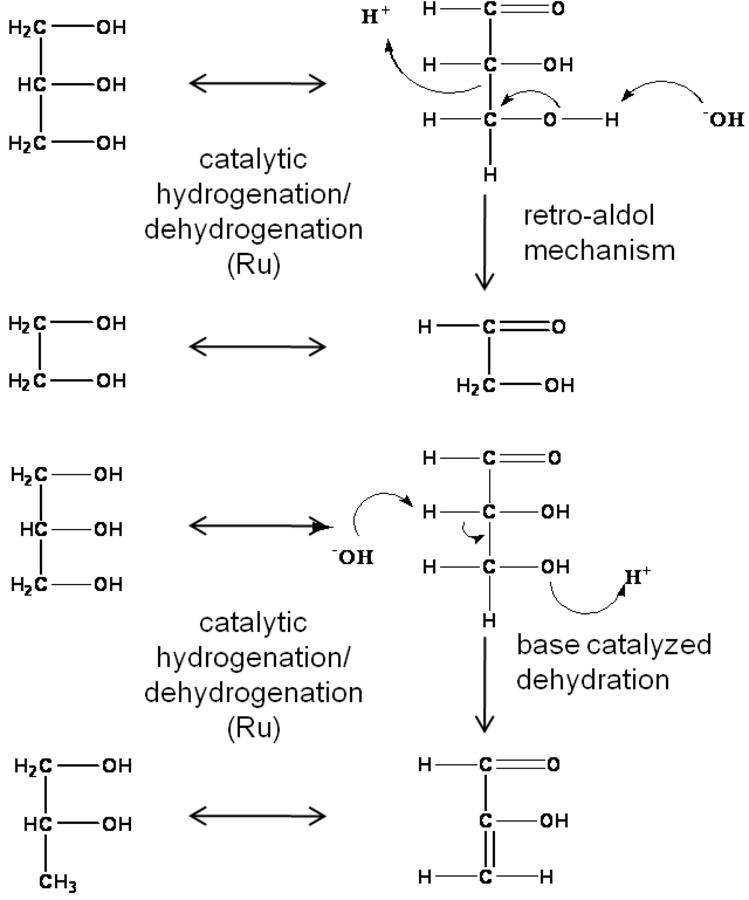
Fig. 11.7: Mechanism that has been proposed for the hydrogenolysis of glycerol to ethylene glycol and propylene glycol
For example, when dehydrating a polyhydroxylated compound it could be a goal to not only control C–O bond cleavage without C–C bond cleavage, but to control which C–O bond is cleaved. A reaction demonstrating these challenges is the hydrogenolysis of alditols with sorbitol, xylitol, and glycerol having received particular attention. This reaction is thought to be initiated by dehydrogenation of the sugar alcohol to either an aldehyde or ketone, so a kinetic mechanism for the system must address the relative reactivity of the hydroxyl groups at the different carbon positions in the molecule as well as the adsorption behavior of the reactant. Hydrogenolysis of alditols provides a useful representative reaction system for the range of reaction considerations that can be encountered in the catalytic conversion of many carbohydrate-derived species, so the reaction can be used to examine the potential of using heterogeneous catalysts for the selective cleavage of C–C and C–O bonds at specific locations within a molecule.
The reaction sequence that has been postulated for the hyddrogenolysis of alditols is shown in Figure 11.7, with glycerol as the reactant. The sequence is thought to be initiated by the dehydrogenation of a hydroxyl group to form either an aldehyde or ketone depending on the hydroxyl group affected [6-8]. This intermediate species can then undergo a C–O bond cleavage through a dehydration reaction, C–C bond cleavage through a retro-aldol reaction, or re-hydrogenation to the initial alditol. The catalyst system most extensively examined for this reaction is Ru on a carbon support, which is used in a highly basic solution. For the previously proposed reaction sequence, the metal function serves to catalyze the dehydrogenation/hydrogenation reactions and the hydroxyls from the basic solution to catalyze the dehydration/retro-aldol reactions. While this straightforward demarcation of catalytic functions for the reaction system is mechanistically attractive, more recent work on glycerol hydrogenolysis has shown that the metal function also plays a role in the cascade of reactions resulting in glycerol dehydration to propylene glycol [2,9,10]. Further complicating the hydrogenolysis reaction, studies with C5 alditols suggest that C–C bond cleavage can potentially occur through decarboxylation as well as through the retro-aldol reaction [4].
While the alditol hydrogenolysis reaction is quite complicated, the fact that the C4 and higher alditols can exist as different stereoisomers (Figure 11.4) can be exploited to gain more insight into the reaction mechanism. For the case of glycerol hydrogenolysis, whether the retro-aldol mechanism (Figure 11.8A) or the decarboxylation mechanism (Figure 11.8B) is the primary path for C–C bond scission cannot be easily resolved as either require formation of glyceraldehyde.
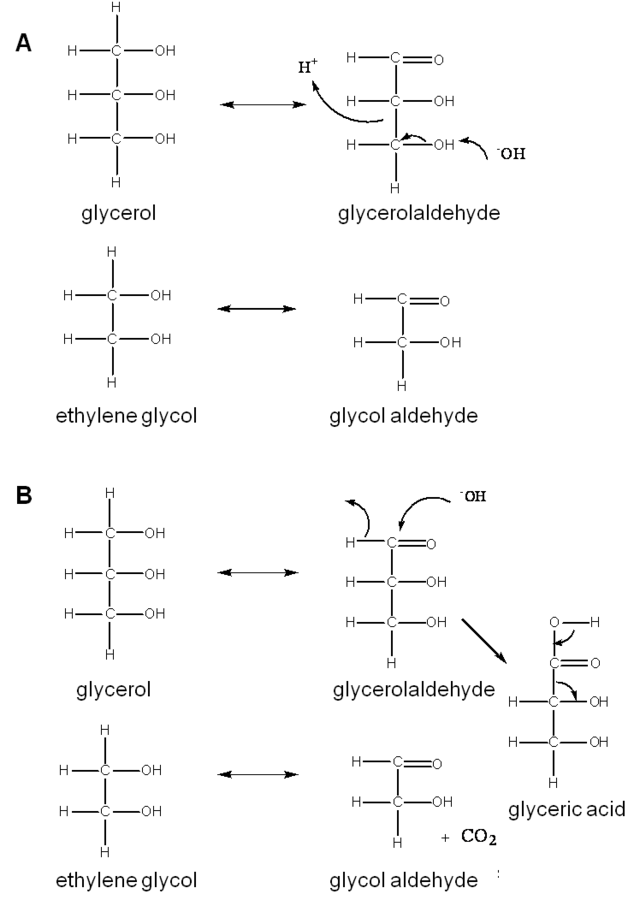
Fig. 11.8: Possible C–C bond scission reactions in polyols: A) retro-aldol reaction, B) decarboxylation reaction
However, consider the case of C–C bond cleavage in xylitol hydrogenolysis where C4 alditols are obtained as products. If decarboxylation was the primary mechanism, the initial step would need to be dehydrogenation of a terminal hydroxyl group (C1 or C5) to yield an aldehyde intermediate. As can be seen from the Fisher Projection of xylitol, cleavage of the C–C bond between either the C1–C2 or C4–C5 would lead to only D-threitol as the product.
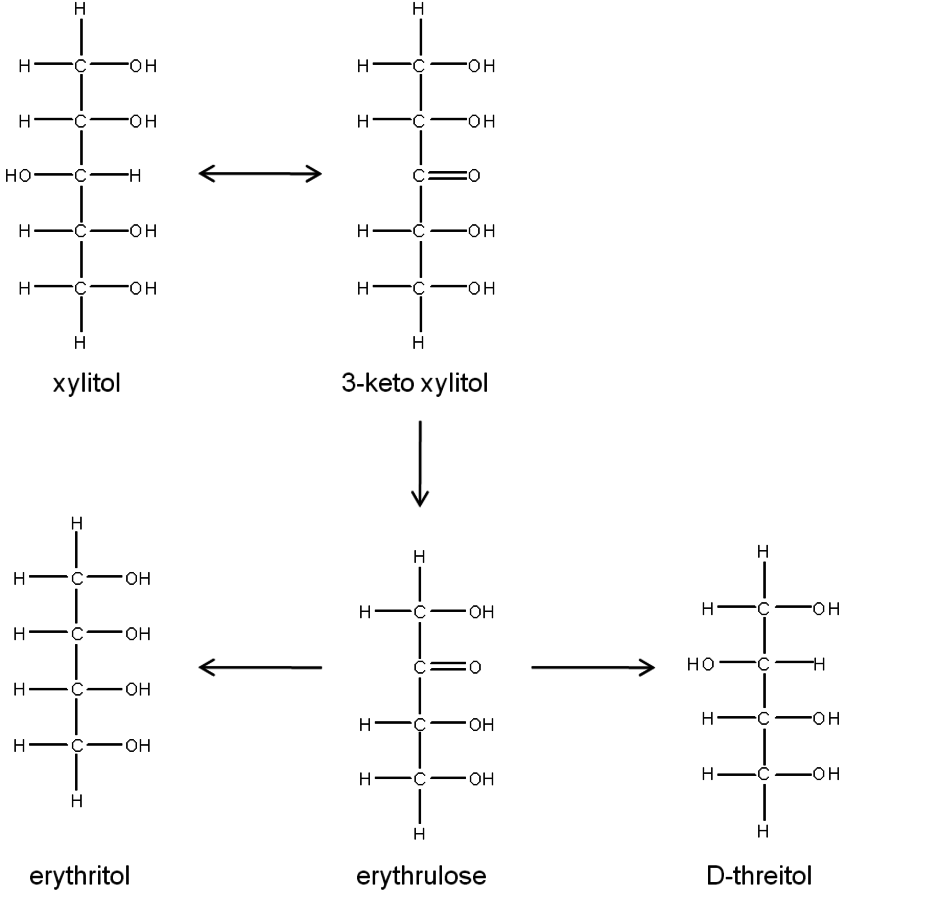
Fig. 11.9: Possible C4 alditols that could result from xylitol when the retro-aldol reaction is initiated by dehydrogenation of the interior hydro-xyl group
The only way in which a C4 alditol could be obtained from xylitol via the retro-aldol reaction would be if the hydroxyl group on the C3 had been dehydrogenated, as dehydrogenation of the hydroxyls at the other carbon positions could only lead to the formation of C2 and C3 polyols. Therefore, the only possibility for C4 alditol formation from xylitol via the retro-aldol mechanism can be shown in Figure 11.9. The first step would be dehydrogenation of the hydroxyl on C3 to form 3-keto xylitol followed by C–C cleavage at C1–C2 or C4–C5, either of which would yield erythrulose. Since the subsequent hydrogenation of the keto group could occur with equal probability from above and below the keto plane, both C4 alditols would be equally likely to form. Therefore, C–C bond scission to C4 alditols via decarboxylation or retro-aldol from xylitol would necessarily yield different product distributions.
Extending from the xylitol example, the possible products resulting from C–C bond scission through hydrogenolysis of C4, C5, and C6 alditols can be mapped as a network of possible products if it is assumed that dehydrogenation of a hydroxyl group is the initiating step. This map is shown in Figure 11.10.
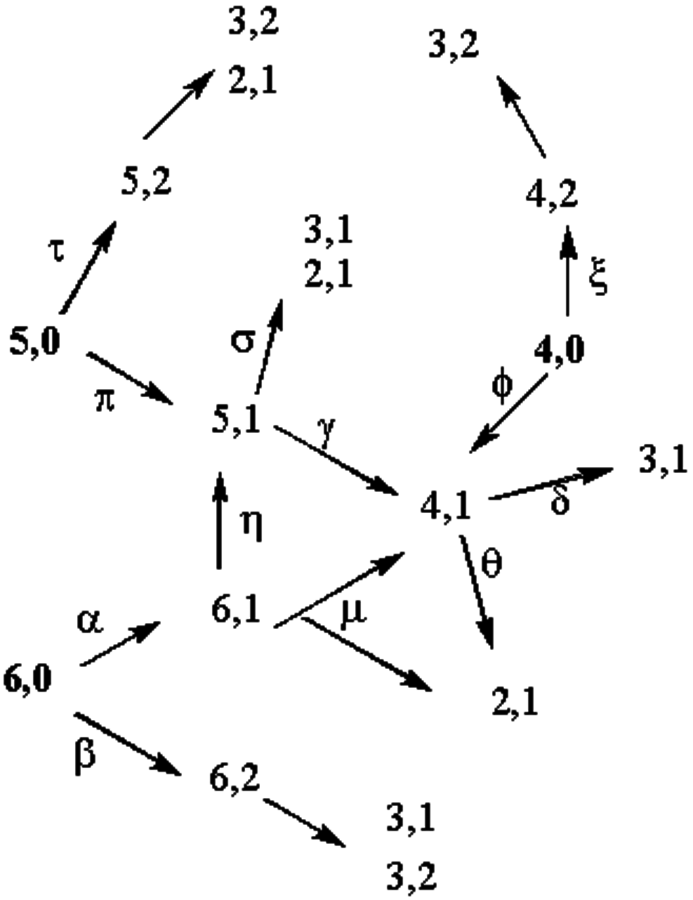
Fig. 11.10: Possible reaction pathways occurring during C–C bond scission in the hydrogenolysis of alditols
To simplify the map visualization, only dehydrogenation of the hydroxyl groups located at the terminal carbons or the carbon one position in from the terminal carbons are shown. In the map, the first number represents the number of carbon atoms in the molecule and the second number the position of the hydroxyl group that is dehydrogenated. For clarification, consider the case of a C5 alditol (5,0). This alditol can dehydrogenate at a terminal carbon to form an aldehyde (5,1), which upon decarboxylation would go to an aldehyde (4,1) or upon retro-aldol reaction to two aldehydes (3,1 and 2,1). Alternatively, the C5 alditol could have dehydrogenation of the hydroxyl group one carbon in from the terminal carbon (5,2), which could only undergo C–C scission via retro-aldol leading to an aldehyde (2,1) and a ketone (3,2). Under the reaction conditions used for hydrogenolysis, the aldehydes and ketones will readily hydrogenate due to the presence of hydrogen and Ru, so only polyhydric alcohols will be observed.
| Temp/(S/Ru) | OAL = 6 | OAL = 5 | OAL = 4 |
|---|---|---|---|
| 205/0 | 0.44 | 0.46 | 0.52 |
| 205/1 | 0.20 | 0.15 | 0.25 |
| 240/0 | 0.44 | 0.37 | 0.59 |
| 240/1 | 0.21 | 0.07 | 0.32 |
Tab. 11.3: Calculated fraction of dehydrogenation taking place on the primary hydroxyl groups based on selectivity mapping of the hydrogenolysis reaction. OAL = Original Alditol Length
Tab. 11.3: Calculated fraction of dehydrogenation taking place on the primary hydroxyl groups based on selectivity mapping of the hydrogenolysis reaction. OAL = Original Alditol Length
Initial conversion studies were performed using most of the C3–C6 alditols [4]. Both their initial rates, as discussed in the previous section, and their reaction products were measured. Importantly, no isomerization was observed from the initial reactant, e.g., when xylitol was used as the reactant no ribitol or arabitol was observed in the product. By combining all of the product distribution data using the mapping approach discussed in conjunction with Figure 11.10, it was possible to calculate whether the hydroxyl group on the terminal carbons were the most readily dehydrogenated under the assumption that only decarboxylation or retro-aldol could give C–C scission. Shown in Table 11.3 are the estimates for the fraction of dehydrogenation occurring at the terminal carbon for two different reaction temperatures (205/0 and 240/0) and three different alditol carbon numbers. As indicated by these results, there is not an equal probability for each of the hydroxyl groups to dehydrogenate as dehydrogenation of the hydroxyl groups on the terminal carbons was somewhat favored.
Several alditol hydrogenolysis studies with Ru on carbon have shown that the addition of sulfur to the reaction system significantly affects the resulting product distribution. For example, sulfur introduction into the glycerol hydrogenolysis reaction system caused the selectivity to propylene glycol to increase relative to ethylene glycol, and its introduction into the xylitol reaction system suppressed the formation of C4 alditols [4,6,9]. To examine this effect, initial conversion studies where performed with the C3-C6 alditols in the presence of sulfur. Shown in Table 11.3 in the 205/1 and 240/1 rows are estimates for the fraction of the dehydrogenation occurring at the terminal carbons when sulfur was added at the level of one mole of sulfur per mole of Ru. These experiments and the analysis mirrored what was discussed in the previous paragraph with the exception of adding sulfur to the reaction system. The addition of sulfur diminished the activity of the catalyst, as reported previously [9], but it also had a large impact on which hydroxyl groups appeared to dehydrogenate most readily. As can be seen by comparison to the 205/0 and 240/0 data, the presence of sulfur appeared to depress dehydrogenation of the hydroxyl groups on the terminal carbons. While the reaction data are phenomenological rather than directly mechanistic, the results clearly demonstrate that the selectivity of bond cleavage can be altered in the hydrogenolysis of alditols with a heterogeneous catalyst.
Several companies have announced the development of commercial processes that are claimed to economically convert either glycerol or sorbitol to propylene glycol. Therefore, the potential exists to cleave selectively C–O and C–C bonds in the presence of multiple hydroxyl groups. However, there is still a need for significantly better understanding of how this cleavage can be achieved selectively and, as such, remains an important research challenge for biorenewable feedstocks.
11.4 Aqueous Phase Considerations
Carbohydrates and their derivatives typically have limited volatility, so their conversion will likely require reactor operation using a condensed phase. As the deconstruction of starch or cellulose to produce monosaccharides is performed in water, subsequent condensed phase processing will likely also involve the aqueous phase. Since heterogeneous catalyst technology has been developed primarily for gas-phase conversions, the determination of how the presence of condensed water at the catalyst/solution interface can influence the kinetics and mechanisms of the resulting reactions has received limited attention. These aqueous phase considerations have implications for both supported metal catalysts as well as solid acid catalysts.
As an example of how the presence of a metal/water interface can influence reaction systems, Neurock and co-workers have used ab initio molecular dynamics to examine several aqueous-phase, metal-catalyzed reaction systems including the dissociation of acetic acid over Pd [11], the hydrogenation of formaldehyde over Pd [12], and the oxidation of CO to
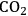 over Pt/Ru [13,14]. Their simulations considered the metal surface and adsorbates as well as an extended ensemble of water molecules. In these computational studies, the presence of the aqueous phase was found to change the thermodynamics and kinetics for many of the elementary reaction steps including adsorption, diffusion, desorption and surface reactions. Perhaps not surprisingly, the reaction steps that involved charge transfer were most strongly affected. One would expect that water would have a solvation effect thereby affecting the thermodynamics and kinetics of the reaction. However, they also found that the condensed phase could play an important role in the reaction mechanism through direct participation in the catalytic pathways.
over Pt/Ru [13,14]. Their simulations considered the metal surface and adsorbates as well as an extended ensemble of water molecules. In these computational studies, the presence of the aqueous phase was found to change the thermodynamics and kinetics for many of the elementary reaction steps including adsorption, diffusion, desorption and surface reactions. Perhaps not surprisingly, the reaction steps that involved charge transfer were most strongly affected. One would expect that water would have a solvation effect thereby affecting the thermodynamics and kinetics of the reaction. However, they also found that the condensed phase could play an important role in the reaction mechanism through direct participation in the catalytic pathways.
One example from their work was the analysis of the possible hydrogenation pathways involved in the Pd-catalyzed hydrogenation of formaldehyde [12], which serves as a potential model for the hydrogenation of the aldehyde groups that could be required in the conversion of carbohydrate-derived species. The presence of the aqueous media was found to reduce the activation barrier for hydrogen addition at either the carbon or oxygen end of the molecule. Perhaps of more interest is that a new energetically favorable pathway enabled by the aqueous phase was found. The new pathway involved a surface hydrogen atom transferring an electron to the surface and then migrating into solution as a proton where the water phase network of local hydrogen bonds facilitated its movement to the active site. The proton then is available to hydrogenate the surface intermediate at the active site. In this new pathway, the metal surface both bonded the adsorbate to the surface and assisted in charge transfer. The charge transfer, which facilitated the electron release from an adsorbed hydrogen creating a proton in solution, was possible because the Pd surface had a work function that was high enough to accept an electron.
The potential for altering a reaction mechanism due to the presence of an aqueous phase was also demonstrated computationally for the dissociation of acetic acid over Pd(111) [11]. When acetic acid was dissociated over Pd in the gas phase, the most energetically favorable pathway was found to be homolytic dissociation with an activation barrier of 468 kJ/mol. Heterolytic dissociation with a computed activation barrier of 1483 kJ/mol did not appear to be possible. When the same reaction system was examined with a condensed aqueous phase the possibility of heterolytic dissociation into an acetate ion and proton was found to be not only energetically attractive with an activation barrier of only 37 kJ/mol, but also favored over homolytic dissociation.
Many reactions of interest in the conversion of biorenewable molecules involve acid catalysis, e.g., hydrolysis, esterification, dehydration, etc., in which a Brønsted acid site is the catalytic moiety. Brønsted acids in aqueous solutions are significantly impacted by solvation through modifying acid strength with the most extreme case being leveling of the acid strength, which occurs when the Brønsted acid sites are completely deprotonated and the acid moiety in the reaction system becomes
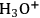 . An additional consideration is that Lewis acid sites when placed in an aqueous medium can be converted to Brønsted acid sites.
. An additional consideration is that Lewis acid sites when placed in an aqueous medium can be converted to Brønsted acid sites.
An example of the effect of proton solvation on catalyst activity was nicely demonstrated in the case of carboxylic acid esterification with methanol [15]. The esterification reaction converts the carboxylic acid and methanol to a methyl ester and water. The condensed phase reaction system used was a starting solution of the carboxylic acid in excess methanol with a solid Brønsted acid catalyst. Studies were also performed in which the starting solution was doped with different low levels of water. It was found that the addition of even small amounts of water into the reacting solution dramatically decreased catalyst activity. This loss in activity was attributed to a decrease in acid strength due to the acidic protons being solvated by the water molecules.
For many reactions catalyzed with Brønsted acids, the activity of the catalyst will correlate with the strength of the acid site, so it is important to be able to measure reliably the pKa values for solid acid catalysts. While reactivity results can be used to indirectly examine acidic site strength, the danger of relying solely on reaction results for characterizing heterogeneous catalysts is that the intrinsic catalytic properties can be obscured due to convolution with mass transfer effects. A number of characterization techniques have been developed to measure independently the strength of acidic sites on solid materials including temperature programmed desorption (TPD), in situ FT-IR and 2-D solid-state NMR, which all primarily make use of adsorbed probe molecules such as carbon monoxide, ammonia, triethylphosphine oxide (TEPO) and pyridine. Unfortunately, these commonly used characterization techniques are based on the interactions of gas-phase probe molecules and cannot account for solvent effects such as proton solvation.
An example of the challenge associated with measuring acid strength for solid acid catalysts to be used in condensed phase reactions is the measurement of the acidic properties of organic acid functionalized materials. A number of studies have examined the acidic properties of propylsulfonic (-
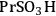 ), ethylphosphonic (-
), ethylphosphonic (-
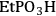 ), butylcarboxylic (-BuCOOH) and arenesulfonic (-
), butylcarboxylic (-BuCOOH) and arenesulfonic (-
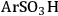 ) functional groups covalently bonded to silica surfaces [16-20]. In a TPD study using pyridine as the probe molecule, the -
) functional groups covalently bonded to silica surfaces [16-20]. In a TPD study using pyridine as the probe molecule, the -
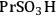 and -
and -
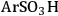 groups were found to have similar acidic strengths with the -
groups were found to have similar acidic strengths with the -
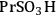 group shown to be a slightly stronger acid [21]. In contrast, a 31P- MAS NMR study with TEPO as the probe molecule indicated that -
group shown to be a slightly stronger acid [21]. In contrast, a 31P- MAS NMR study with TEPO as the probe molecule indicated that -
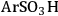 was the stronger acidic moeity [20]. This discrepancy was also seen in other studies, where the acidic trends among the functional groups were dependent on the method employed and/or the probe molecule used. Additionally, the solvent used in the reaction system can affect the acidic strength of the catalytic sites. A study comparing the acid strength of sulfonated polystyrene resin and sulfonated mesoporous silica in both aqueous and non-aqueous media [22] found molar enthalpies of neutralization suggesting that the sulfonated polystyrene material was more acidic than the sulfonic-acid functionalized silica material in water, but the result was found to be the opposite in a non-aqueous media.
was the stronger acidic moeity [20]. This discrepancy was also seen in other studies, where the acidic trends among the functional groups were dependent on the method employed and/or the probe molecule used. Additionally, the solvent used in the reaction system can affect the acidic strength of the catalytic sites. A study comparing the acid strength of sulfonated polystyrene resin and sulfonated mesoporous silica in both aqueous and non-aqueous media [22] found molar enthalpies of neutralization suggesting that the sulfonated polystyrene material was more acidic than the sulfonic-acid functionalized silica material in water, but the result was found to be the opposite in a non-aqueous media.
A computational study was performed to compare how the acidic properties for organic acid functionalized silica were impacted by the presence of a single water molecule versus several water molecules [23]. This comparison began to examine how acidic strength can be influenced by the presence of a single probe molecule relative to an ensemble of molecules, which is in the direction of a condensed phase calculation. In this work, the interaction of the -
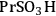 , -
, -
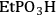 , -BuCOOH, -
, -BuCOOH, -
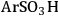 functional groups with water was examined using the Effective Fragment Potential method with density functional theory (DFT). The simulations were performed for the cases in which the functional groups were bound to a silicon atom that was either capped with hydroxyl groups or linked to an oxygen-silicon structure. The optimized structures for each of the functional groups and silicon (capped using the two different models) and one water molecule is shown in Figure 11.11.
functional groups with water was examined using the Effective Fragment Potential method with density functional theory (DFT). The simulations were performed for the cases in which the functional groups were bound to a silicon atom that was either capped with hydroxyl groups or linked to an oxygen-silicon structure. The optimized structures for each of the functional groups and silicon (capped using the two different models) and one water molecule is shown in Figure 11.11.
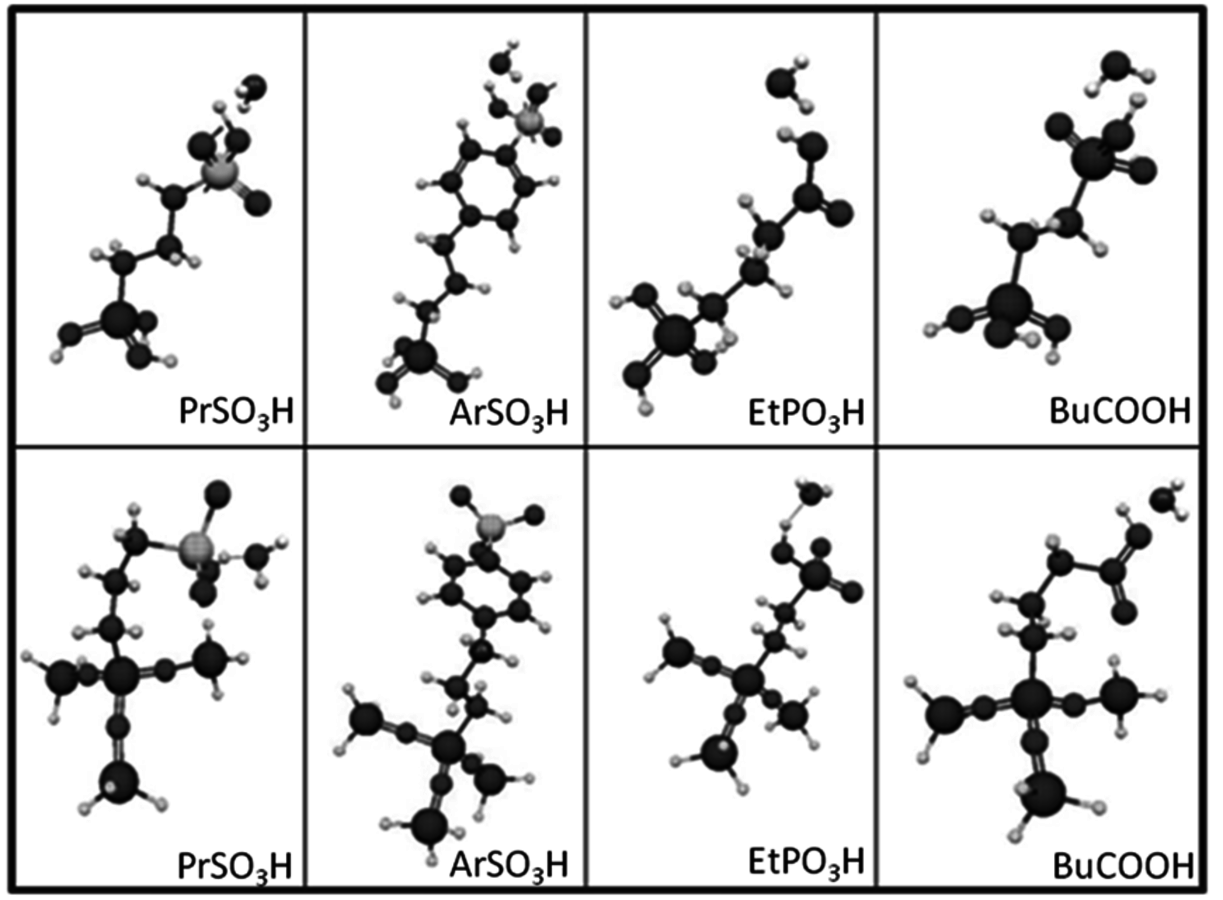
Fig. 11.11: DFT optimized acid functional group structures with one water molecule; top row with silanol groups and bottom row without
The elongation of the O-H bond in the acidic moiety has been used in literature to predict pKa values with greater elongation of this bond correlated with lower pKa values [24]. Shown in the first row in Table 11.4 is the O-H bond distance for each of the tethered functional groups without water present. The second row gives the elongated bond length after a single water molecule was introduced into the computation. The elongated bond length was calculated in a ratio with the initial bond length to get the percent elongation for the O-H bond due to a single water molecule. As can be seen from the table, the computational results suggested that acidic strength of the functional groups followed the trend of -
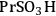 > -
> -
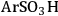 > -
> -
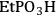 > -BuCOOH.
> -BuCOOH.
| PrSO3H w/ SiOH |
PrSO3H w/o SiOH |
PhSO3H w/ SiOH |
PhSO3H w/o SiOH |
|
|---|---|---|---|---|
| O–H bond distance (Å) | 0.9761 | 0.9706 | 0.9700 | 0.9629 |
| elongated O–H distance (Å) | 1.0455 | 1.0384 | 1.0381 | 1.0299 |
| elongation % | 7.11 | 6.99 | 7.02 | 6.96 |
| EtPO3H w/ SiOH |
EtPO3H w/o SiOH |
BuCOOH w/ SiOH |
BuCOOH w/o SiOH |
|
|---|---|---|---|---|
| O–H bond distance (Å) | 0.9689 | 0.9661 | 0.9584 | 0.9570 |
| elongated O–H distance (Å) | 1.0204 | 1.0159 | 1.0027 | 1.0002 |
| elongation % | 5.32 | 5.16 | 4.62 | 4.51 |
Tab. 11.4: Calculated bond distances and O–H bond elongation resulting from the interaction of a single water molecule with the functional group
Tab. 11.4: Calculated bond distances and O–H bond elongation resulting from the interaction of a single water molecule with the functional group
The trend predicted for the interaction of the different organic acid functional groups did not follow that observed from aqueous phase titration of the acids, which gave an acid strength ordering of -
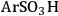 > -
> -
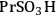 > -
> -
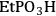 >
>
-BuCOOH [25]. Therefore, the computational work was extended by adding water molecules up to a total of four with the optimized structures (minimized energy) shown in Figure 11.12. While four molecules were still considerably short of representing a continuous condensed phase, the O-H bond elongation percent was calculated. As shown in Table 11.5, the relative ordering of the O-H bond length elongation for these four functional groups now followed the same pKa trend that was observed from experimental titrations with the -
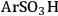 group having the longest O-H bond elongation. These results demonstrate the challenge associated with characterizing Brønsted acid catalysts relative to their use in the aqueous phase, or other solvent phases, as traditional acid site characterization techniques might not adequately represent how the catalytic moiety might be expected to perform under condensed phase conditions.
group having the longest O-H bond elongation. These results demonstrate the challenge associated with characterizing Brønsted acid catalysts relative to their use in the aqueous phase, or other solvent phases, as traditional acid site characterization techniques might not adequately represent how the catalytic moiety might be expected to perform under condensed phase conditions.
Fig. 11.12: DFT optimized acid functional group structures with four water molecules
| PrSO3H | PhSO3H | EtPO3H | BuCOOH | |
|---|---|---|---|---|
| O–H bond distance (Å) | 0.9761 | 0.9700 | 0.9689 | 09584 |
| elongated O–H distance (Å) | 1.0555 | 10553 | 1.0225 | 1.0077 |
| elongation % | 8.13 | 8.79 | 5.53 | 5.15 |
Tab. 11.5: Calculated bond distances and O-H bond elongation resulting from the interaction of four water molecules with the functional group in the presence of silanol groups.
Tab. 11.5: Calculated bond distances and O-H bond elongation resulting from the interaction of four water molecules with the functional group in the presence of silanol groups.
Overall, the presence of an aqueous phase in a catalytic reaction system dictates that catalyst characterization approaches developed for catalysts used in gas-phase reactions need to be carefully considered when applied to catalysts that are to be used in aqueous-phase applications. Additionally, there is a need to develop new methodologies that are appropriate for characterizing how catalysts might be expected to perform in the presence of an aqueous solvent.
11.5 Novel Catalytic Materials for Biomass Conversion
It is reasonable to expect that catalyst materials that were developed for gas-phase applications with hydrocarbons will not likely be the best catalysts for condensed phase applications with biomass-derived molecules. While there is only a limited precedent for translating standard heterogeneous catalysts into their effective use for converting biorenewable feedstocks, there are catalyst systems that have developed to be efficient in this application. Enzymes have evolved to be quite active and selective in the conversion of biorenewable molecules with an important aspect being the creation of a reaction sphere around the active site. These active domains in enzymes consist of amino acid residue groupings in which the amino acid residues surrounding the specific active site amino acid residue can modify the reaction sphere thereby modifying the reactivity and selectivity of the active site.
Shown in Figure 11.13 is a rendering of a cellobiohydrolase enzyme, which is active for hydrolyzing the glycosidic bond in a cellobiose molecule [26]. It is interesting to see how this enzyme has evolved to create an active and selective catalytic domain. First, the enzyme has a folded tunnel that creates a hydrogen bonding gradient, which can effectively “pull” the cellobiose substrate to the active domain within the enzyme. When reaching the active domain, the cellobiose substrate docks into a configuration that places torsional pressure on the glycosidic bond, thereby lowering its activation barrier for the hydrolysis reaction. Within the active domain there are two amino acid residues that act in concert to catalyze the reaction. Figure 11.14 shows the machanism thought to be occurring in this enzyme for the hydrolysis reaction (known as the inverting mechanism). In the inverting mechanism, a deprotonated amino acid residue activates a water molecule by nucleophilic attack, which activates the water to interact with carbon next to the glycosidic bond. Since the water molecule is donating electron density to this carbon, the glycosidic C–O bond is weakened sufficiently such that a proton from a carboxylic acid group on another amino acid residue can more easily break the bond. The activation barrier for glycosidic bond cleavage with a typical cellobiohydrolase enzyme is about 35 kJ/mol compared to about 120 kJ/mol with a mineral acid ([27]), which demonstrates how an enzyme can dramatically modify reaction activation barriers by controlling the entire reaction domain.
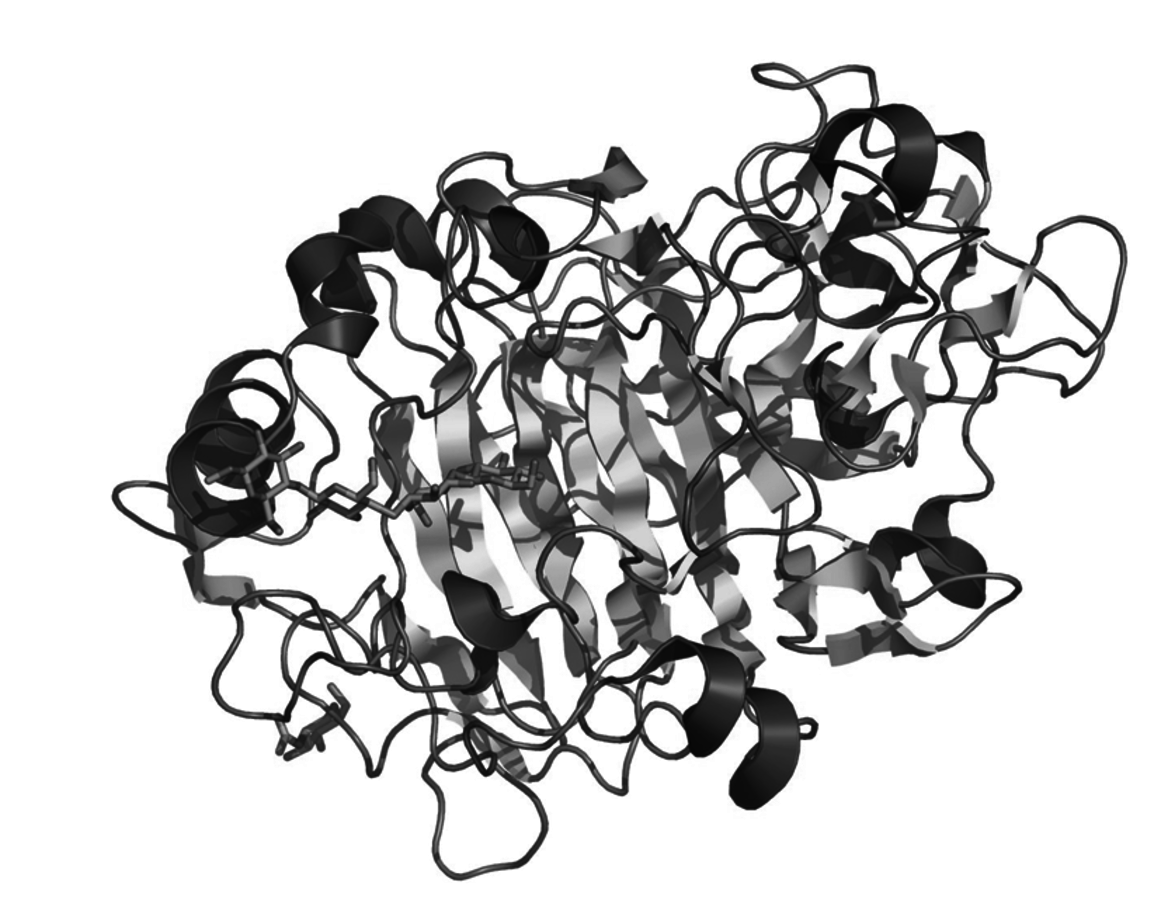
Fig. 11.13: Representation of a glycoside hydrolase, family 7 cellobiohydrolase, with the cellobiose substrate entering the reactive domain
Examination of the cellobiohydrolase enzyme shows that the enzyme controls the reaction domain by providing preferential access of the desired substrate to the active site, adsorbing the substrate in a form that activates the compound for the reaction, and providing cooperative action between two catalytic sites for improved activity. Taking this cue from nature, catalytic materials in which both the active site and the broader reaction domain are carefully controlled may prove to be types of materials that are particularly effective for the active and selective conversion of biorenewables. In gas-phase reactions, there is precedent for heterogeneous materials that have some of the same elements of reaction domain control, but the catalytic requirements for these reaction domains when used in converting biorenewable feedstocks in the condensed phase will undoubtedly be different.
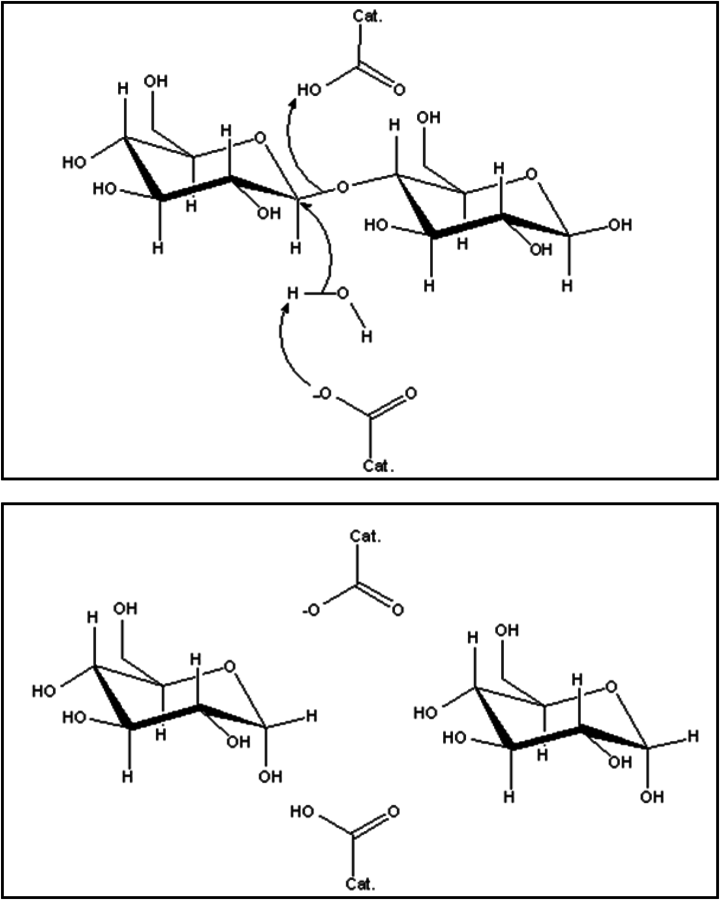
Fig. 11.14: Proposed inverting mechanism reaction for the hydrolysis of a β-1,4 glycosidic bond with a hydrolase enzyme
The esterification reaction with biorenewable feedstocks has been the subject of several studies focused on improving catalytic performance by controlling the reaction environment in solid materials, Diaz et al. [28,29] examined glycerol esterification with fatty acids using organic acid-functionalized mesoporous silicas. The goal of the reaction was preferentially to produce monoglycerides while minimizing subsequent esterification reactions, which lead to the formation of di- or triglycerides. They used three attributes within the mesoporous materials to control their catalytic performance. First, propylsulfonic acid groups were tethered to the interior of the pores within the mesoporous materials.
Mesoporous silica was used as the support so that only a narrow pore size distribution existed. Additionally, the average pore diameter of these pores was controlled by choice of the surfactant template used in the synthesis of the mesoporous support. By using a strong acid group in conjunction with a tuned pore diameter, a MCM-41-
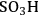 catalyst having both high activity and selectivity for glycerol esterification to monoglycerides only was identified. The good catalytic performance of the propylsulfonic acid-functionalized mesoporous silica catalyst was attributed to a combination of strong acidic sites located inside the mesopores and to the narrow pore size distribution, which diminished the formation of secondary di- and triglyceride products. The catalytic activity of the propylsulfonic acid-functionalized mesoporous silica catalyst was compared to that of a homogeneous acid (p-toluenesulfonic acid). The homogeneous acid had high activity but poor selectivity to the desired monosaccharide. A third attribute that was examined was the introduction of a second organic group within the pore through functionalizing MCM-41 materials with alkyl groups as well as propylsulfonic acid groups. It was found that the esterification reaction rate was improved with an increase in alkyl group loading, which was speculated to be due to water being excluded from the reaction environment within the pore. The methylated propylsulfonic acid-functionalized MCM-41 catalyst had a three-fold increase in catalytic activity relative to the non-methylated organosulfonic acid-functionalized mesoporous silica material.
catalyst having both high activity and selectivity for glycerol esterification to monoglycerides only was identified. The good catalytic performance of the propylsulfonic acid-functionalized mesoporous silica catalyst was attributed to a combination of strong acidic sites located inside the mesopores and to the narrow pore size distribution, which diminished the formation of secondary di- and triglyceride products. The catalytic activity of the propylsulfonic acid-functionalized mesoporous silica catalyst was compared to that of a homogeneous acid (p-toluenesulfonic acid). The homogeneous acid had high activity but poor selectivity to the desired monosaccharide. A third attribute that was examined was the introduction of a second organic group within the pore through functionalizing MCM-41 materials with alkyl groups as well as propylsulfonic acid groups. It was found that the esterification reaction rate was improved with an increase in alkyl group loading, which was speculated to be due to water being excluded from the reaction environment within the pore. The methylated propylsulfonic acid-functionalized MCM-41 catalyst had a three-fold increase in catalytic activity relative to the non-methylated organosulfonic acid-functionalized mesoporous silica material.
Mesoporous scaffolds, having uniform sized pores, offer the opportunity to control the reaction environment around an active site throughout the catalytic material by controlling the placement of functional groups within the pores and/or on the exterior of the metal oxide particles. Controlled placement of functional groups in and on mesoporous silicas was examined, with the resulting catalysts tested in the esterification of fatty acids with methanol [30-32]. The esterification reaction, which produces an alkyl ester and water as the reaction product, is reversible. While the equilibrium constant at typical esterification reaction temperatures strongly favors the products over the fatty acid and methanol reactants, the constraint imposed by equilibrium becomes important at high conversion levels of the reactants. For organosulfonic acid functionalized mesoporous silicas, the tethered functional groups within the pores are surrounded by silanol groups, so the pores are hydrophilic. As it is desirable to exclude water from the catalytically active domain, a material having hydrophobic pores would be preferred. A set of catalytic materials were synthesized to control the hydrophilicity/hydrophobicity of the pores in mesoporous particles by introducing tethered alkyl groups.
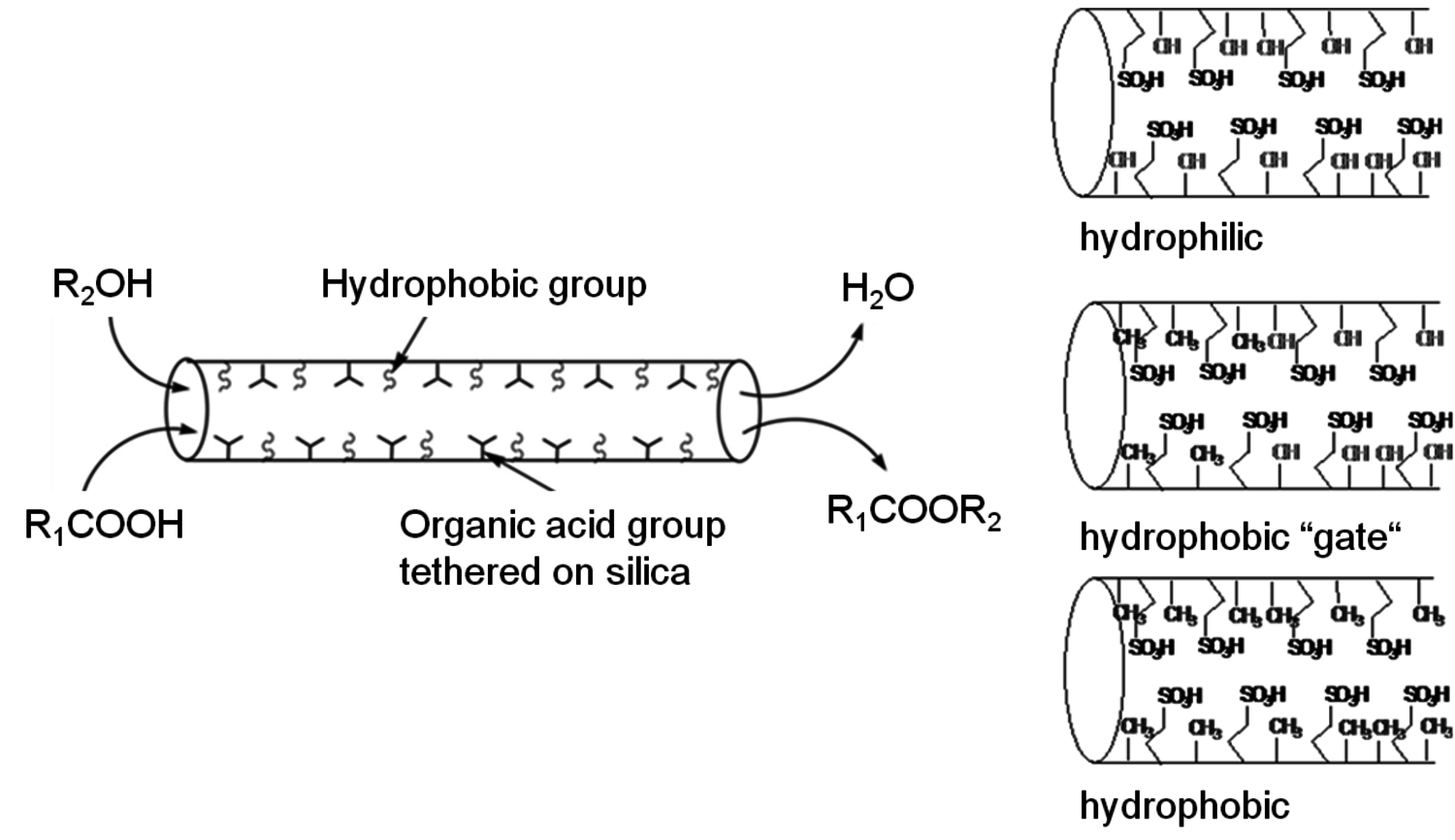
Fig. 11.15: Schematics of propylsulfonic acid functionalized mesoporous silicas with different incorporation of alkyl groups either no incorporation (top), post-grafted (middle), and co-condensed (bottom)
Shown in Figure 11.15 are idealized schematics of the materials resulting from three different synthesis approaches [30]. Hydrophilic pores result from co-condensation of organosulfonic acid and silica precursors in the presence of a surfactant template. Studies have shown that the co-condensation synthesis creates materials in which all of the tethered functional groups reside on the interior of the pores. This material would represent the most hydrophilic case. Grafting an alkyl group onto these sulfonic acid functionalized silicas via condensation of an alkyl alkoxysilane precursor with the surface silanols will result in tethering the alkyl group to the external surface of the particles as well as to the pore mouth since the silanol groups at these locations are the most readily accessible. This material will effectively have a hydrophobic “gate” as the external surface of the particle and entrance to the pore will still be hydrophobic, while most of the interior of the pore will still be hydrophilic. Alternatively, the organosulfonic acid, alkyl, and silica precursors can all be co-condensed leading to a material that has a more hydrophobic pore. The mesoporous silica catalyst that was synthesized by co-condensing all of the precursors, which placed the hydrophobic groups within the pores, gave significantly higher catalytic performance than the catalyst without hydrophobic group incorporation and the grafted catalyst, which would preferentially place the hydrophobic groups on the particle surfaces and near the pore mouths [30].
Another interaction effect exploited by enzymes is the interaction between multiple amino acid residues, which can modify the acid strength of the residue that directly serves as the active site. This cooperativity effect can be seen with heterogenous catalysts as well as it has been shown that the location of propylsulfonic acid groups within a mesoporous material can alter the acidity of the resulting material. Intentionally forcing sulfonic acid groups to be in close proximity through the use of a disulfide precursor decreased the pKa value relative to the same number of acidic sites incorporated using a propyl thiol precursor, which would not force two sulfonic acid groups to be in close proximity (see schematic in Figure 11.16) [19,32].
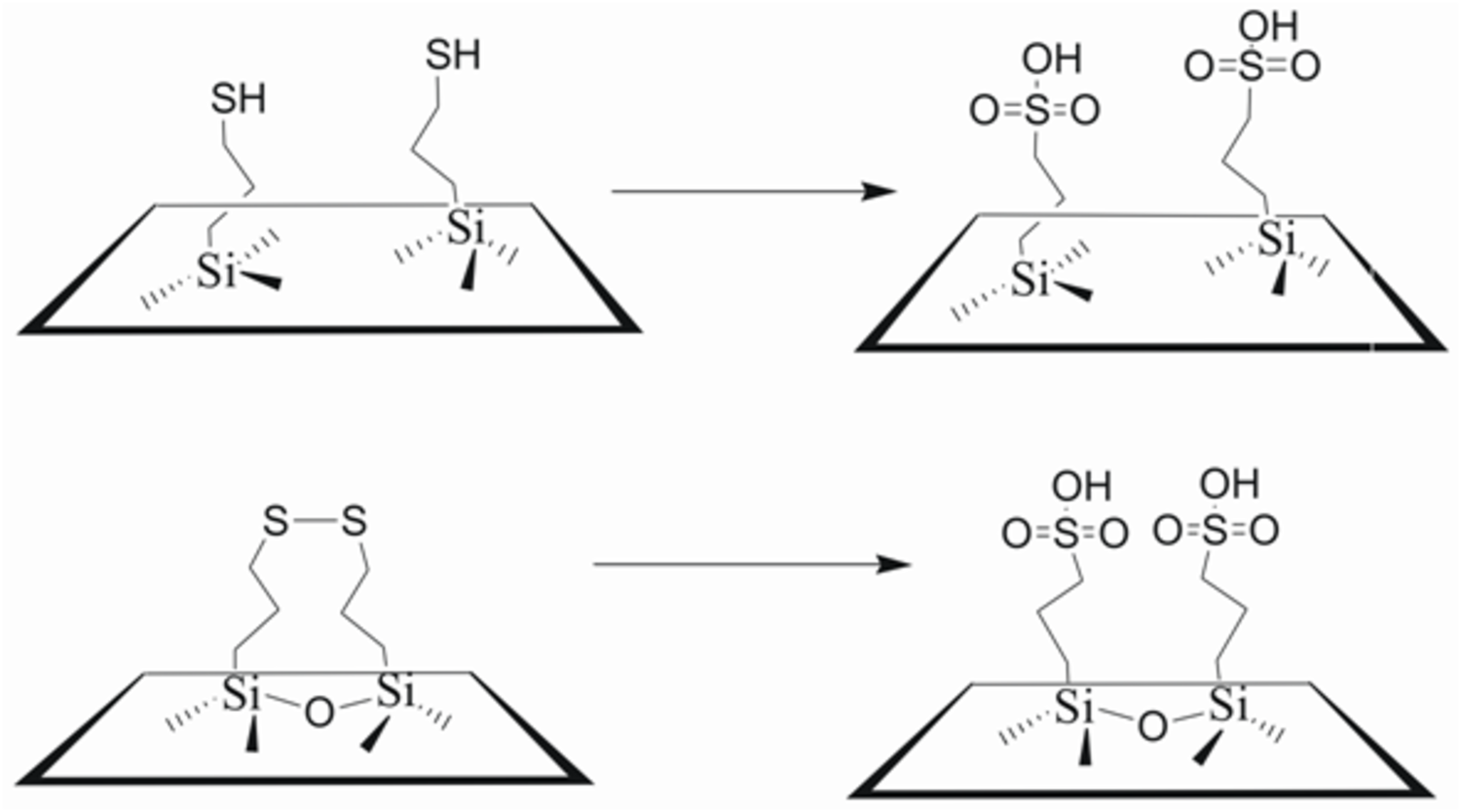
Fig. 11.16: Controlled spatial placement of propylsulfonic acid groups through the use of a propyl thiol precursor (top) or a propyl disulfide precursor (bottom)
Enhanced reactivity due to cooperative effects between catalytic moieties can also be seen for cases in which two different catalytic moieties are interacting as shown in a study involving a bifunctional catalyst with both strong acid and metal sites [33]. One method to deconstruct solid biomass into smaller molecular species existing in the condensed phase is the fast pyrolysis process. Fast pyrolysis generates a “bio-oil” that is a mixture of over 200 chemical species resulting from the thermal deconstruction of lignocellulose. While these bio-oils hold potential for upgrading to fuels, they are too acidic and unstable. Aldehydes and organic acids in the bio-oil are important contributors to the instability and acidity, so their removal would be desirable. In principle, the aldehydes and organic acids could be simultaneously removed by hydrogenating the aldehydes to alcohols, which could then be esterified with the organic acids to form esters. Metal catalysis is required for the hydrogenation step and acid catalysis for the esterification reaction. This concept is shown schematically in Figure 11.17.
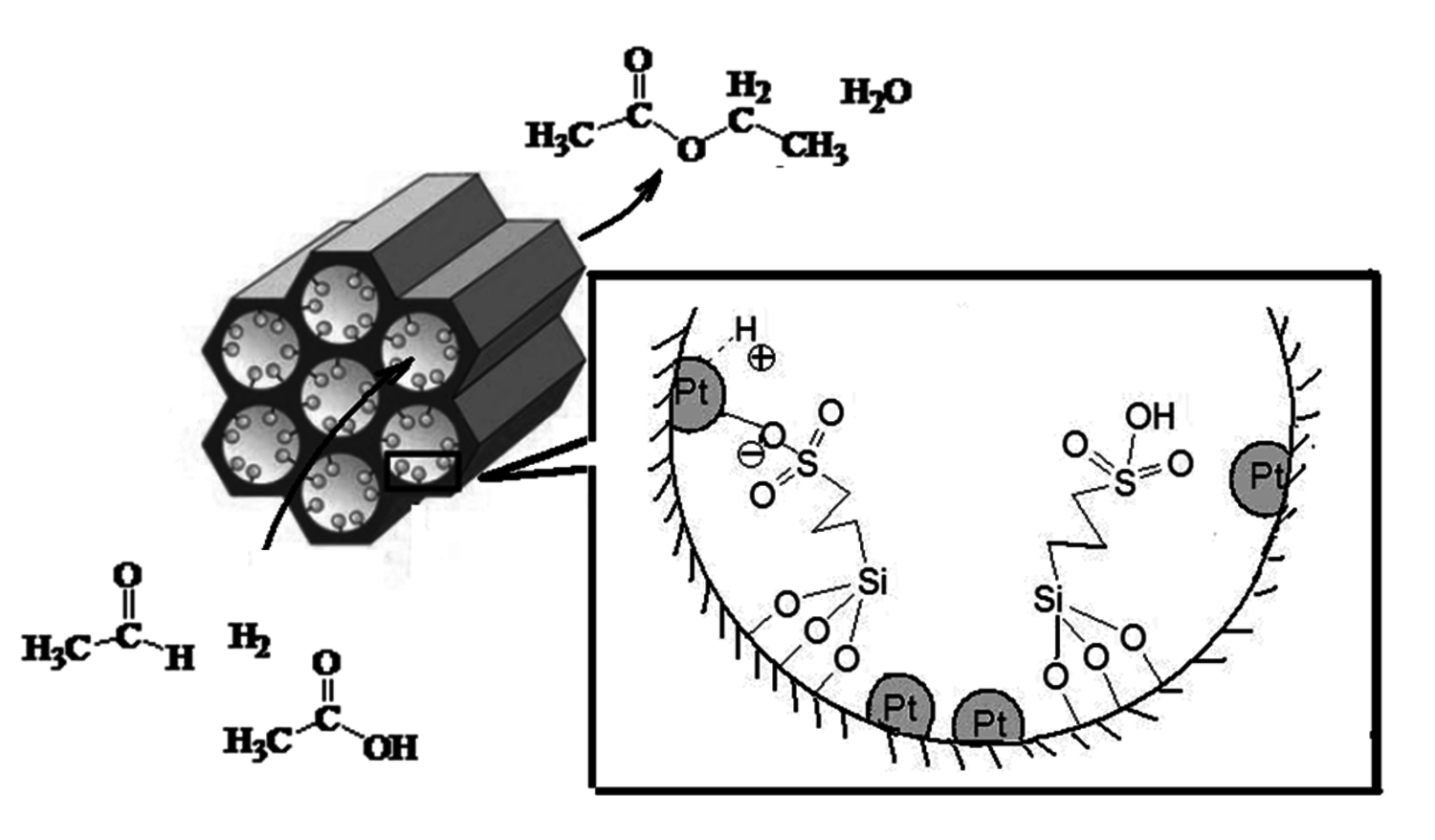
Fig. 11.17: Schematic of the bifunctional catalyst used in the simultaneous hydrogenation and esterification of aldehydes and organic acids
To accomplish combined hydrogenation/esterification a mesoporous catalyst was synthesized in which propylsulfonic acid groups were tethered to the interior pores through co-condensation followed by incorporation of Pt through reductive deposition [33]. A TEM image of the resulting bifunctional catalyst is shown in Figure 11.18. Upon titration characterization of the bifunctional catalysts, the presence of Pt was found to lower the pKa of the acidic groups as shown in Table 11.6, where the pKa of the propylsulfonic acid functionalized silica was 3.91, which was subsequently lowered to 3.55 and 3.31 when nominal loadings of 1 wt% and 5 wt% Pt were incorporated, respectively. Shown in Table 11.7 are comparative reactivity results for the monofunctional catalyst and the bifunctional catalysts. As can be seen from the first column the hydrogenation activity was only dependent on whether Pt was present on the catalyst as the monofunctional Pt and bifunctional catalysts had the same activities. However, the bifunctional catalyst had superior esterification activity than the monofunctional propylsulfonic acid catalyst as well as a physical mixture of the monofunctional Pt and proplylsulfonic acid catalysts. These results were in complete agreement with the acid strength measurements and confirmed that the Pt and propylsulfonic acid groups had to be in close proximity to achieve the cooperative acid strength effect.

Fig. 11.18: TEM images of Pt impregnated on propylsulfonic acid functionalized mesoporous silica
| Catalysts | Acid sites mmol/g | pKa |
|---|---|---|
| SBA15-PrSO3H | 0.89 | 3.91 |
| Pt(1)/SBA15-PrSO3H | 0.69 | 3.55 |
| Pt(5)/SBA15-PrSO3H | 0.57 | 3.31 |
Tab. 11.6: Number of acid sites and pKa values for propylsulfonic acid functionalized mesoporous silicas upon incorporation of Pt
Tab. 11.6: Number of acid sites and pKa values for propylsulfonic acid functionalized mesoporous silicas upon incorporation of Pt
| Catalyst | Hydrogenation (CH3CHO conv., 24 h) | Esterification (TON of HAc) | Esterification (HAc conv., 24 h) |
|---|---|---|---|
| SBA15-PrSO3H | – | 3000 | – |
| Pt/SBA15 | 8.1% | – | – |
| PtSBA15-PrSO3H | 7.9% | 8000 | 8.4% |
| Physical mixture: SBA15-PrSO3H+Pt/SBA15 | – | – | 3.5% |
Tab. 11.7: Activity comparison for a series of mesoporous silica catalysts with various functional groups (150 °C and 15 bar H2)
Tab. 11.7: Activity comparison for a series of mesoporous silica catalysts with various functional groups (150 °C and 15 bar H2)
11.6 Hydrothermally Stable Catalytic Materials
Reaction systems in the condensed phase create a new challenge involving developing catalysts that possess adequate physical stability. This challenge is magnified when the reaction is being performed in the aqueous phase. Hydrothermal stability of heterogeneous catalysts has received a great deal of attention as it is a longstanding issue for typical metal oxide based catalysts. Even so, much of the work on creating hydrothermally stable solid catalysts has focused on their recalcitrance to water vapor and not condensed water. Aqueous-phase catalytic conversions of biorenewables are demanding as they will necessitate solid catalysts that can maintain their stability to temperatures as high as 230 °C.
An attractive feature of metal oxide supports is that they provide high surface area to disperse catalytic species and can be readily functionalized due to the presence of surface hydroxyls. Unfortunately, both of these characteristics diminish the hydrothermal stability of the metal oxides as high surface area coupled with high surface hydroxyl content makes the metal oxides more easily hydrolyzed. One approach to improving hydrothermal stability is to calcine the metal oxides at elevated temperatures, which will remove surface hydroxyls and decrease surface area. This effect can be seen in Figure 11.19, where mesoporous SBA-15 silicas were tested for hydrothermal stability in liquid water at 145 °C for 2 hours. Shown on the left in the figure is the pore size distribution of the mesoporous silicas, as synthesized and calcined (500 °C for 1 hour), prior to hydrothermal treatment. The overall pore volume diminished slightly upon calcination, but the average pore diameter remained essentially the same. After the hydrothermal treatment, the pore structure of the uncalcined mesoporous silica completely collapsed while the calcined material maintained its structure. When the hydrothermal treatment was performed at 175 °C in water for 2 hours, even the structure of the calcined mesoporous silica collapsed.
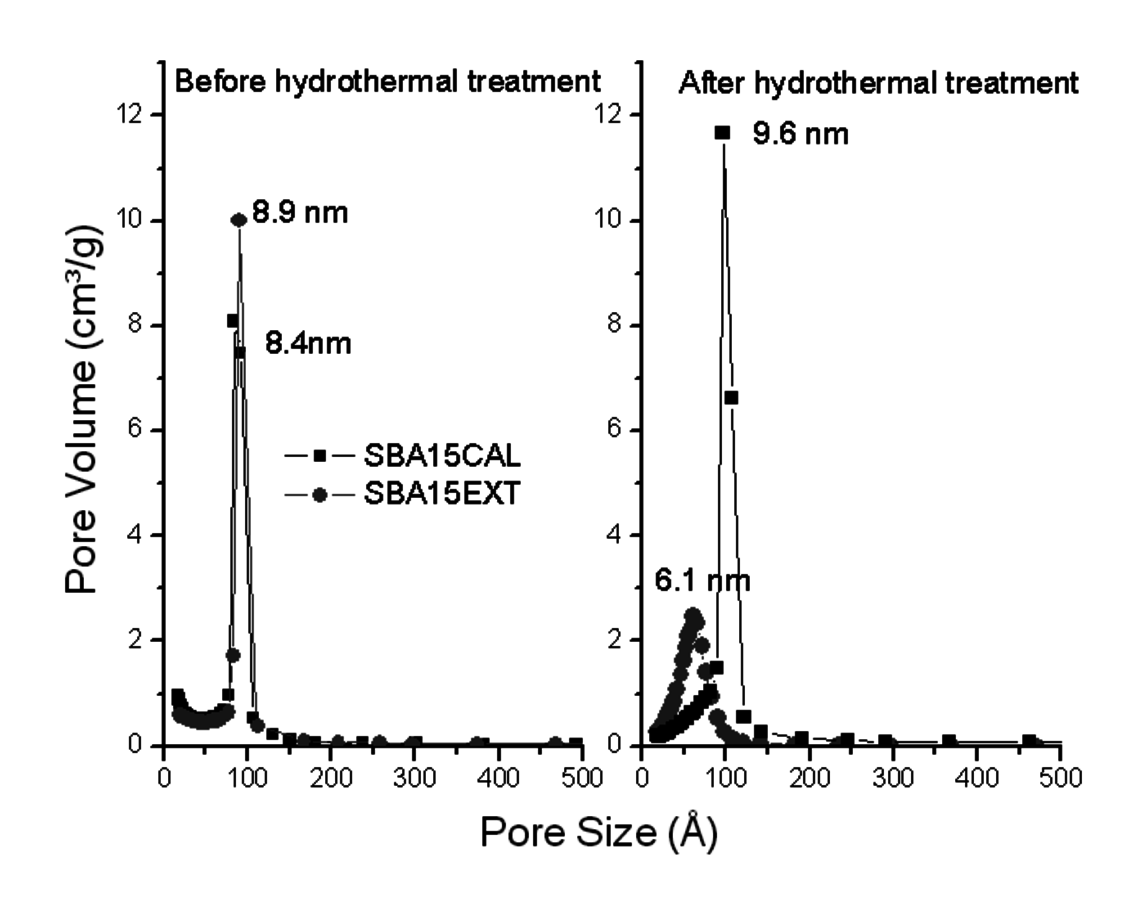
Fig. 11.19: Pore size distribution of SBA-15 as synthesized and after calcination with subsequent hydrothermal treatment
As discussed in previous sections, many transformations of carbohydrates and their derivatives will require the use of Brønsted acid catalysts. Unfortunately, acidic aqueous solutions further exacerbate the hydrothermal stability problem as the acidic proton can catalyze the hydrolysis of metal oxides. When a calcined mesoporous silica was subjected to hydrothermal treatment at 145 °C in water for 2 hours with the water at pH4, the pore structure of the silica was found to collapse. Therefore, metal oxides are not promising supports for use in aqueous reactions at elevated temperatures particularly if acid catalysis is desired.
An interesting class of acidic materials that has been examined for use in the conversion of biorenewable molecules is sulfonated carbons. Using glucose or sucrose carbonization followed by sulfonation with concentrated sulfuric acid, sulfonated carbons were produced [34,35]. Depending on the carbonization conditions, carbon-based materials with acid content of up to 1.34 mmol/g were reported. It was speculated that the carbonized carbohydrate formed polycyclic aromatic sheets with the sulfonic acid groups bound to the edges of the sheets [34]. The sulfonated carbon materials were found to have good activity for fatty acid esterification with methanol and were stable to repeated batch reaction testing at temperatures of 80–180 °C. However, it should be noted that the water content was fairly low as the only water present was that produced in the esterification reaction.
In a more rigorous test of the stability of sulfonated carbons in aqueous media, the materials were tested using the cellulose hydrolysis reaction at 150 °C [34]. For this study, the sulfonated carbon catalysts were synthesized by sulfonating activated carbon rather than carbonized carbohydrates. While the catalyst was found to maintain its activity through three hydrolysis cycles, an independent measure of the number of sulfonic acid groups remaining at the end of the three cycles was not performed to determine whether sulfur loss had occurred.
While using sulfonated carbonized carbohydrates appeared to yield catalysts with good stability in condensed phase reactions, these materials were not easily amenable for producing materials in which the pore size distribution could be controlled. Therefore, several alternative synthesis approaches were explored for creating mesoporous sulfonated carbons with narrow and tunable pore size distributions. In one approach, sucrose was impregnated onto preformed SBA-15 and then the combined material was carbonized [36]. After carbonization, the original silica framework was removed using HF washing and the resulting mesoporous structured carbon was sulfonated. The carbon-based catalytic material was reported to be stable when used for multiple reaction cycles in the liquid-phase Beckmann rearrangement of cyclohexanone oxime. A similar approach was used to make sulfonated carbon-silica mesoporous composites in which the silica was not removed [37]. The MCM-48-based composite materials were found to maintain their ordered mesoporous structure upon being exposed to boiling water for 48 hours, whereas the MCM-48 mesoporous structure when not coated with carbon collapsed within 3 hours when exposed to boiling water. Additionally, the sulfonated carbon-silica material was found to be an active esterification catalyst.
The studies with the sulfonated carbon materials appear to suggest that these materials have promising hydrothermal stability properties. However, key characterization validating the long-term stability of the materials has still not been reported. While reproducible activity through 3–5 batch reaction cycles is important, the limited number of cycles is inadequate to determine whether the sulfonic acid groups are still slowly being lost. Catalyst activity is an indirect measure of sulfonic acid group content so small losses of active sites can be obscured by other reaction factors such as reactant and product measurement accuracy, etc. Therefore, it is desirable to have an independent measure of the number of active sites retained. Additionally, acid-catalyzed biorenewable reactions can require that the catalysts be stable in the aqueous phase at temperatures as high as 230 °C, so the sulfonated carbon materials need to be tested at temperatures above boiling water to determine their potential range of application.
| Sample | Acidic sites mmol H+/g |
Sample II | Acidic sites mmol H+/g |
|---|---|---|---|
| SC0 | 1.21 | SC0 | 1.37 |
| SC1 | 1.18 | SC1 | 1.28 |
| SC2 | 1.08 | SC2 | 1.22 |
| SC3 | 1.07 | ||
| SC(175) | 1.10 |
Tab. 11.8: Stability of acid sites on sulfonated carbon subjected to hydrothermal treatment in liquid water (145 °C for 2 hours in each treatment)
Tab. 11.8: Stability of acid sites on sulfonated carbon subjected to hydrothermal treatment in liquid water (145 °C for 2 hours in each treatment)
Shown in Table 11.8 and Figure 11.20 are results for the stability of a sulfonated carbon catalyst, which was made by sulfonation of carbonized sucrose following the method of Toda et al. [35]. The fresh sulfonated carbon sample was found via titration to have an acid site content of 1.37 mmol
 /g. The starting material was split into 5 samples, which were subjected to different hydrothermal treatment conditions. Sample SC0 had no hydrothermal treatment, while SC1, SC2, and SC3 were subjected to 1, 2, and 3 hydrothermal treatment cycles, respectively, at 145 °C in water for 2 hours. SC(175) had one hydrothermal treatment at 175 °C in water for 2 hours. Shown in Figure 11.20 is the performance of these sulfonated carbon materials in the acetic acid esterification reaction with methanol. This reaction was used to evaluate the catalysts as it has been found to be quite sensitive to the number of acidic groups and the strength of the acidic groups. Within the experimental error, SC0, SC1, and SC2 all appeared to have comparable activities from the reaction studies, but as can be seen from Table 11.8 there was a slight loss of acidic sites from SC0 to SC1 to SC2. A large decrease in activity was observed for SC3 and SC(175), which was accompanied with a significant decrease in the number of active sites. This example is illustrative of the risk associated with characterizing catalyst stability only according to its performance under reaction conditions as the reaction results would suggest stability for SC0, SC1, and SC2 when more complete characterization demonstrated that the materials were not ultimately stable for extended use.
/g. The starting material was split into 5 samples, which were subjected to different hydrothermal treatment conditions. Sample SC0 had no hydrothermal treatment, while SC1, SC2, and SC3 were subjected to 1, 2, and 3 hydrothermal treatment cycles, respectively, at 145 °C in water for 2 hours. SC(175) had one hydrothermal treatment at 175 °C in water for 2 hours. Shown in Figure 11.20 is the performance of these sulfonated carbon materials in the acetic acid esterification reaction with methanol. This reaction was used to evaluate the catalysts as it has been found to be quite sensitive to the number of acidic groups and the strength of the acidic groups. Within the experimental error, SC0, SC1, and SC2 all appeared to have comparable activities from the reaction studies, but as can be seen from Table 11.8 there was a slight loss of acidic sites from SC0 to SC1 to SC2. A large decrease in activity was observed for SC3 and SC(175), which was accompanied with a significant decrease in the number of active sites. This example is illustrative of the risk associated with characterizing catalyst stability only according to its performance under reaction conditions as the reaction results would suggest stability for SC0, SC1, and SC2 when more complete characterization demonstrated that the materials were not ultimately stable for extended use.
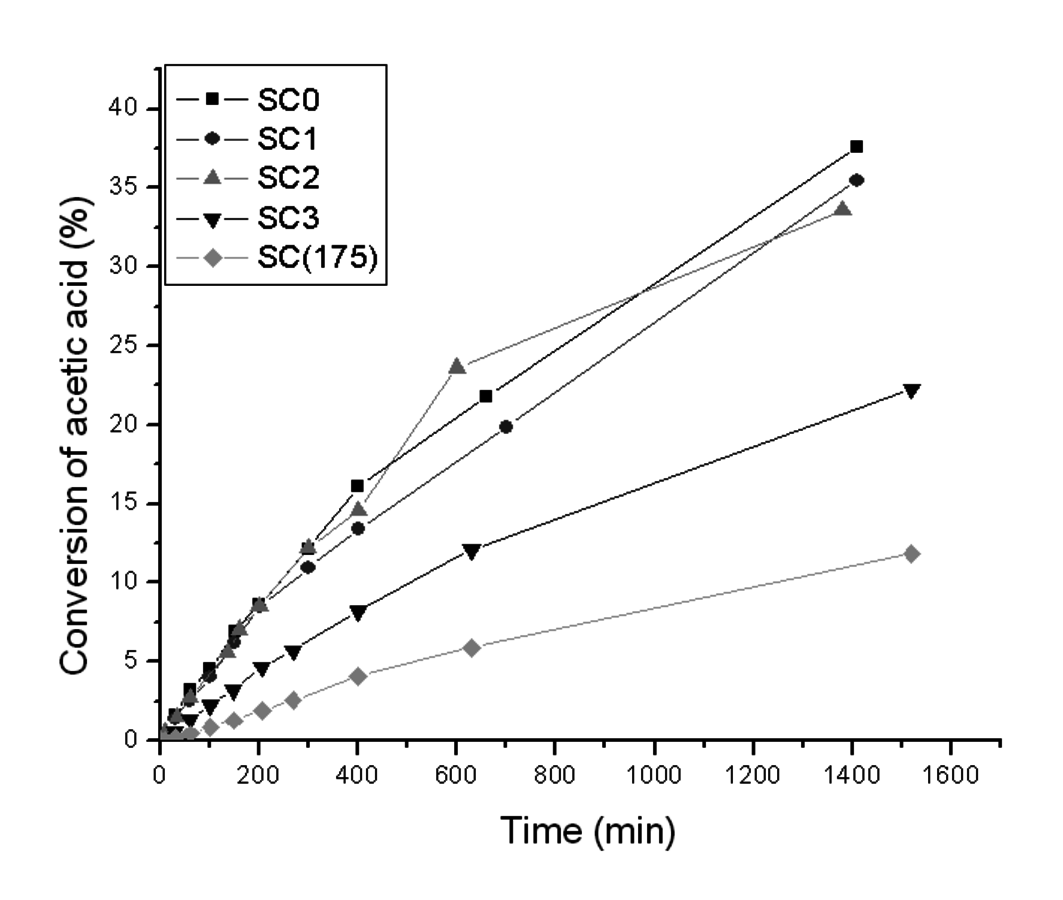
Fig. 11.20: Esterification activity of sulfonated carbons after hydrothermal treatment
Finding solid catalysts that are stable under condensed phase hydrothermal conditions remains an important challenge. The challenge is more significant when attempting to synthesize a solid Brønsted acid catalyst, as the acidity has an adverse affect on stability. In exploring hydrothermal stability of solid catalysts it is important to ensure that the characterization techniques being used are truly representative of determining whether the catalytic material is hydrothermally stable for the prolonged use needed in a real catalytic process.
11.7 Impurity Tolerant Catalysts
Frequently, the development of catalysts for converting new feedstocks focuses on how to accomplish the transformation of these new reactants to the desired products. There is no doubt that demonstrating successful transformation chemistry is vital when evaluating new types of feedstocks. However, new feedstocks can introduce the additional issue of low-level impurities that are intrinsic to that feedstock. For example, the primary low-level impurities of concern in processing crude oil are sulfur and nitrogen heteroatoms. Biorenewable feedstocks introduce a new array of low-level impurities that can have significant impact on heterogeneous catalyst performance.
Triglyceride transesterification and fatty acid esterification to alkyl esters, which are used as biodiesel, have received a great deal of attention. The primary catalysts used commercially in these conversions, sodium methoxide for transesterification and sulfuric acid for esterification, are homogeneous, which necessitates a downstream catalyst removal step. There has been a large number of papers examining possible heterogeneous catalysts capable of replacing the homogeneous catalysts. While catalyst activity and stability against leaching of the active sites are critical properties for the heterogeneous catalysts, dealing with impurities in the feedstocks will also play a role in developing viable heterogeneous catalyst systems.
An example demonstrating the importance of impurities for alkyl esters production is the esterification of fatty acids in lipid feedstocks containing elevated levels of free fatty acids, i.e., a portion of the triglycerides have been hydrolyzed creating fatty acids that are not bound up as esters. Typically, the first step in processing such a feedstock is to use a homogenous acid, commonly sulfuric acid, to esterify the free fatty acids to alkyl esters, with the remaining triglycerides then transesterified with a strong base catalyst. Using model feedstocks in which 15 wt% of a fatty acid was doped into purified triglycerides, it was shown that an organosulfonic acid functionalized mesoporous silica catalyst was an effective replacement for sulfuric acid in the esterification reaction [38]. However, when the solid catalyst was used for esterifying free fatty acids in a real feedstock, beef tallow with 7 wt% free fatty acid, the catalyst deactivated quite rapidly [31]. In successive batch reactor runs with the same catalyst, the catalyst activity was halved from the first cycle to the second cycle and then halved again in the third cycle (see Figure 11.21A). The most likely source of this deactivation was adsorption of polar compounds, such as phospholipids or proteins, that are known to exist at low levels in unpurified lipid feedstocks. A simple purification of the beef tallow accomplished by passing it over a bed of activated silica, which had a highly hydroxylated surface that would provide for adsorption of polar compounds, dramatically improved the performance of the catalyst, as now the activity of the solid esterification catalyst decreased by only about 15% from the first cycle to the second. Although an improvement, the activity loss was still too high for the catalyst to be of practical relevance. Therefore, a catalytic material was made in which organosulfonic acid and alkyl precursors were co-condensed with the silica precursor leading to mesoporous silica with tethered organosulfonic acid and alkyl groups (see schematic in Figure 11.21B). The alkyl groups were introduced to make the surface more hydrophobic and thereby less likely to adsorb polar compounds.
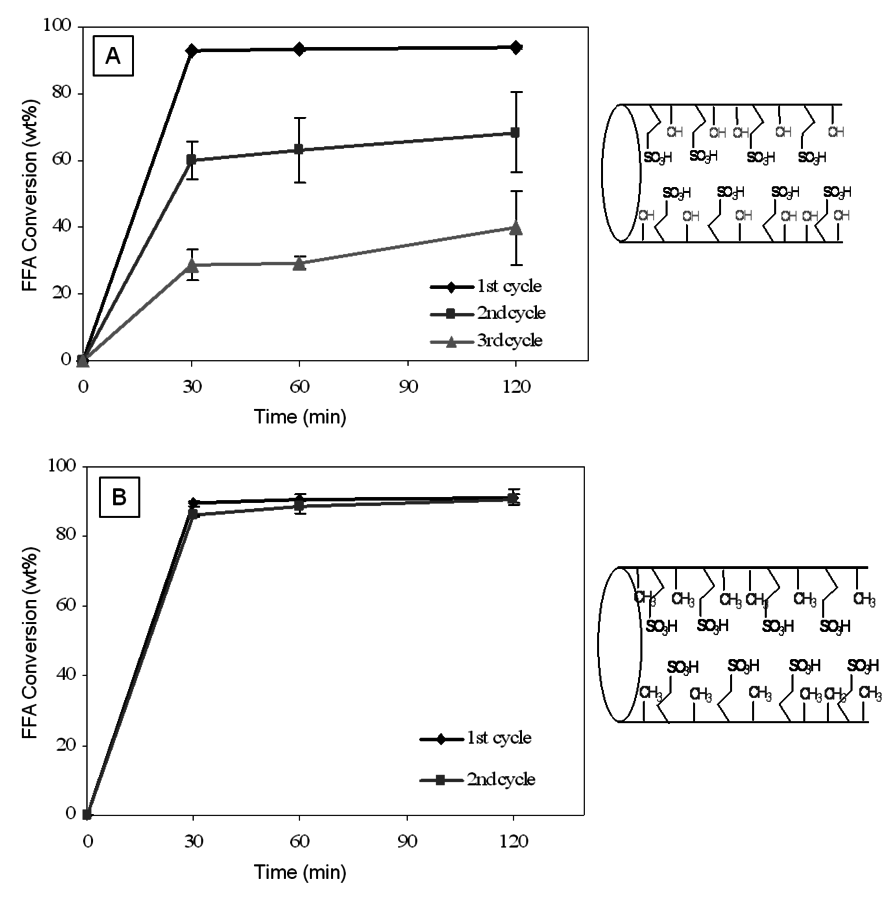
Fig. 11.21: Performance of propylsulfonic acid mesoporous silica in the conversion of beef tallow: A) loss of activity when the catalysts is used in multiple cycles, B) activity of resulting when incorporating alkyl groups in the pores and pretreating the feed with activated silica
Shown in Figure 11.21B is the performance of the catalyst system in which the activated silica was used prior to the alkyl-modified organosulfonic acid functionalized mesoporous silica catalyst. As can be seen in the figure, this combination led to a catalyst system in which no loss of activity was observed between the first and second cycles. While two cycles were insufficient to demonstrate long term stability, a clear improvement in performance was realized.
Another type of impurity that is present to varying levels in lignocellulosic feedstocks is metal ions. Shown in Table 11.9 are metal ions commonly present in lignocellulosic biomass. Included in the table are the most common forms in which the metal ions exist, which includes both as salts and as organically associated species [39]. A more quantitative determination of impurity content in a lignocellulosic feedstock is shown in Table 11.10 [40]. For these data, switchgrass was combusted and the resulting ash analyzed for its metal oxide content. Of particular importance for the use of heterogeneous catalysts with biomass is the significant presence of alkalis and alkaline earths, which are known to be poisons for many types of metal catalysts. Therefore, catalysts must be developed that can either tolerate these impurities or a pretreatment system needs to be employed that can remove the impurities prior to exposure to the catalyst. This issue becomes important in processes in which lignocellulosic biomass is thermally deconstructed such as by gasification or pyrolysis with subsequent catalytic upgrading to desired products. In general, pretreatment to remove alkalis and alkaline earths from biomass is quite expensive, so there will be a need to deal with these species downstream from the gasification or pyrolysis step.
| Element | Compound | Share of the total element (%) |
|---|---|---|
| Ionic form, water soluble | ||
| Na | NaCl, NaNO3 | > 90 |
| K | KCl, KNO3 | > 90 |
| Mg | Mg(NO3)2, MgCl2, Mg3(PO4)2 | 60–90 |
| Ca | Ca(NO3)2, CaCl2, Ca3(PO4)2 | 20–60 |
| Organically Associated | ||
| Mg | Chlorophyll | 8–35 |
| Ca | Ca-oxalate, Ca-pectate | 30–85 |
| Mn, Fe | Other macromolecules | > 80 |
Tab. 11.9: Typical forms of metal ions in biomass materials
Tab. 11.9: Typical forms of metal ions in biomass materials
| Compound | Content (wt%) |
|---|---|
| SiO2 | 72.2 |
| Al2O3 | 3.74 |
| Fe2O3 | 1.28 |
| SO3 | 0.39 |
| CaO | 6.19 |
| MgO | 2.33 |
| Na2O | 0.77 |
| K2O | 6.17 |
| P2O5 | 2.63 |
| TiO2 | 0.18 |
| SrO | 0.02 |
| BaO | 0.01 |
Tab. 11.10: Composition of the ash resulting from the combustion of switchgrass
Tab. 11.10: Composition of the ash resulting from the combustion of switchgrass
11.8 Novel Reaction Systems
As reaction systems for biorenewables will frequently involve a condensed phase, separation processes will also need to be developed that can purify the desired products from the reaction medium. To improve the economic viability of biorenewable conversions, it is useful to design catalysts in concert with reaction systems such that the subsequent separation can be facilitated. While the goal of integrating separation considerations with the reaction system typically involves producing a product that can be more easily removed from the reaction medium, another objective can be to enhance product selectivity. The multifunctional nature of biorenewable-derived molecules makes selective conversion to a desired product more difficult since the product will often have significant reactivity as well. Therefore, removal of the product from the reaction medium can have the advantage of improved selectivity beyond what might be possible just from catalyst modifications.
One example of integrating reaction and separation is work from the Dumesic lab that examined the conversion of aqueous-phase carbohydrates to monofunctional hydrocarbons (alcohols, ketones, heterocyclics, and acids) using a Pt-Re catalyst [41,42]. In this work, C4 and higher monfunctional species were produced that were sufficiently hydrophobic such that they formed an organic phase, which was then immiscible from the reactant-containing aqueous phase. Starting with a 60 wt% solution of sorbitol in water, they were able to recover as much as
Fig. 11.22: Schematic of biphasic reactor system for converting fructose to HMF [43]
60 mol% of the original feedstock carbon within an organic phase using a reaction system that was operating at 230 °C, 27 bar and a space velocity of 1.2 hr-1 [41]. Therefore, a simple separation of the product was achieved by operating the system at conditions leading to the formation of hydrophobic monofunctional hydrocarbons. The monofunctional hydrocarbons could then be directly upgraded to fuels through aldol condensation and ketonization C–C coupling reactions to produce fuels without any further intermediate separation [44-48].
A second example in which separation considerations were effectively incorporated into the design of a biorenewable reaction system was the biphasic production of hydroxymethylfurfural (HMF) from fructose [43,49,50]. It is difficult to achieve high selectivity in the dehydration of fructose to HMF, since the acid catalyst needed for dehydration is also quite active for the further reaction of HMF to undesirable products. By using a biphasic reaction system (see schematic in Figure 11.22), the reactive aqueous phase, which contained the fructose feedstock and the acid catalyst, was immiscible with an extracting phase, which contained an organic solvent having high HMF solubility. The extraction of the HMF to the nonreactive organic phase greatly reduced the subsequent conversion of HMF, which both increased the yield to the desired product and increased the fructose conversion. By appropriate selection of the extracting solvent and the addition of a salt or phase modifiers (DMSO and 1-methyl-2-pyrrolidinone) to the aqueous phase to further improve the partitioning of HMF into the extracting phase, high selectivities to the HMF product were achieved. For example, a 30 wt% fructose solution saturated with NaCl using 1-butanol as the extracting solvent gave an HMF selectivity of 82% whereas in a simple single phase reaction the HMF selectivity was only about 28%.
11.9 Summary
Over the past several years there has been a rapid acceleration of studies aimed at understanding how to tailor heterogeneous catalysts and the reaction systems utilizing them for the efficient conversion of biorenewable feedstocks with a number of them given in this chapter. It is clear that the catalytic materials needed to convert successfully biorenewable feedstocks must address reaction characteristics that are distinctly different from those encountered when developing catalytic materials for the refining and petrochemical industry. While catalyst development for biorenewables can build from knowledge for hydrocarbon conversions, there is a need to develop evaluation techniques that can adequately address the new attributes imposed through this alternative feedstock, which have been discussed in the current chapter. The economic hurdle to replace fossil carbon with renewable carbon in chemical and fuel applications is quite demanding, so significant improvements in catalyst technology are required. To achieve the necessary heterogeneous catalyst advancements, more advanced fundamental knowledge about how catalysts can be designed specifically for converting biorenewable feedstocks is still needed and, as such, the unique challenges associated with designing catalysts for this application will be a rich source of new research directions for heterogeneous catalysis.
Bibliography
[1] L. Peereboom, J.E. Jackson, J. J.. Interaction of Polyols with Ruthenium Metal Surfaces in Aqueous Solution Green Chemistry 11: 1979-1986 (2009): 1979-1986.
[2] D.G. Lahr, B.H. Shanks. Kinetic Analysis of the Hydrogenolysis of Lower Polyhydric Alcohols: Glycerol to Glycols Industrial & Engineering Chemistry Research 42: 5467 (2003): 5467.
[3] M.K. Koshikawa, T. Hori. Adsorption Selectivity of Sugars toward Hydrous Zirconium(IV) and Hydrous Iron(III) Oxide Surfaces Physical Chemistry Chemical Physics 2: 1497-1502 (2000): 1497-1502.
[4] D.G. Lahr, B.H. Shanks. Effect of Sulfur and Temperature on Ruthe- nium-Catalyzed Glycerol Hydrogenolysis to Glycols Journal of Catalysis 232: 386 (2005): 386.
[5] R.J. Ferrier. Carbohydrate Chemistry. Monosaccharides, Disaccharides, and Specific Oligosaccharides Carbohydrate Chemistry 26: 1 (1994): 1.
[6] C. Montassier, J.C. Menezo, J. M., Hoang J.C., H. J.. Aqueous Polyol Conversions on Ruthenium and on Sulfur-Modified Ruthenium Journal of Molecular Catalysis 70: 99 (1991): 99.
[7] E. Tronconi, N. Ferlazzo, N. F., Forzatti N., F. N., P. Forzatti. A Mathematical Model for the Hydrogenolysis of Carbohydrates Chemical Engineering Science 47: 2451-2356 (1992): 2451-2356.
[8] K. Wang, M.C. Hawley, M. H.. Mechanism Study of Sugar and Sugar Alcohol Hydrogenolysis Using 1,3-Diol Model Compounds Industrial & Engineering Chemistry Research 34: 3766 (1995): 3766.
[9] D.G. Lahr. Mechanistic Insight into the Production of Ethylene Glycol and Propylene Glycol from Biorenewable Resources. phdthesis. Iowa State University, 2005
[10] E.P. Maris, R.J. Davis. Hydrogenolysis of Glycerol over Carbon-Sup- ported Ru and Pt Catalysts Journal of Catalysis 249: 328-337 (2007): 328-337.
[11] S.K. Desai, P. Venkataraman, P. V.. A Periodic Density Functional Theory Analysis of the Effect of Water Molecules on Deprotonation of Acetic Acid over Pd(111) Journal of Physical Chemistry B 105: 9171-9182 (2001): 9171-9182.
[12] S.K. Desai. Theoretical Investigations of Solution Effects on Metal Catalyzed Hydrogenation and Oxidation Processes. phdthesis. University of Virginia, 2002
[13] S.K. Desai, M. Neurock. A first Principles Analysis of CO Oxidation over Pt and Pt66.7 Ru33.3 (111) Surfaces Electrochimica Acta 48: 3759-3773 (2003): 3759-3773.
[14] S.K. Desai, M. Neurock. First-Principles Study of the Role of Solvent in the Dissociation of Water over a Pt-Ru Alloys Physical Review B 68: 075420 (2003): 075420.
[15] E. Lotero, Y. Liu, Y. L., Lopez Y., L. Y., D.E. Lopez. Synthesis of Biodiesel via Acid Catalysis Industrial & Engineering Chemistry Research 44: 5353-5363 (2005): 5353-5363.
[16] S. Hamoudi, S. Royer, S. R.. Propyl- and Arene-Sulfonic Acid Functionalized Periodic Mesoporous Organo Silicas Microporous and Me- soporous Materials 71: 17-25 (2004): 17-25.
[17] V. Herledan-Semmer, S. Dakhlaoui, S. D., Guenneau S., G. S.. Spectroscopic Evidence of SBA-15 Mesoporous Solids Functionalization Studies in Surface Science and Catalysis 154: 1519-1524 (2004): 1519-1524.
[18] S. Koujout, D.R. Brown. Calorimetric Base Adsorption and Neutralization Studies of Supported Sulfonic Acids Thermochimica Acta 434: 158-164 (2005): 158-164.
[19] R.K. Zeidan, V. Dufaud, V. D.. Enhanced Cooperative, Catalytic Behavior of Organic Functional Groups by Immobilization Journal of Catalysis 239: 299-306 (2006): 299-306.
[20] R. Kanthasamy, I.K. Mbaraka, I. M., Shanks I.K.. Solid-State MAS NMR Studies of Sulfonic Acid-Functionalized SBA-15 Applied Magnetic Resonance 32: 513-526 (2007): 513-526.
[21] S. Hamoudi, D.T. On, D. O.. Mesostructured Solid Acids Studies in Surface Science and Catalysis 14: 15-22 (2003): 15-22.
[22] S. Koujout, B.M. Kiernan, B. K., Brown B.M., B. B., D.R. Brown. The Nature of the Internal Acid Solution in Sulfonated Poly(sty- rene-co-divinylbenzene) Resins Catalysis Letters 85: 33-40 (2003): 33-40.
[23] B. Cinlar. Acid Catalyzed Carbohydrate Degradation and Dehydration. phdthesis. Iowa State University, 2010
[24] P. Lorente, I.G. Shenderovich, I. S., Golubev I.G., G. I., N.S. Golubev. 1H/15N NMR Chemical Shielding, Dipolar 15N,2H Coupling and Hydrogen Bond Geometry Correlations in a Novel Series of Hydrogen-Bonded Acid-Base Complexes of Collidine with Carboxylic Acids Magnetic Resonance in Chemistry 39: 18-29 (2001): 18-29.
[25] B. Cinlar, B.H. Shanks. Characterization of the Acidic Sites in Organic Acid Functionalized Mesoporous Silica in the Aqueous Phase Applied Catalysis A: General (in press):.
[26] P. Reilly. Courtesy of Professor Peter Reilly, Iowa State University.. .
[27] J.A. Bootsma, B.H. Shanks. Cellobiose Hydrolysis Using Organic-Inorganic Hybrid Mesoporous Silica Catalysts Applied Catalysis A: General 327: 44-51 (2007): 44-51.
[28] J. Perez-Pariente, I. Diaz, I. D., Mohino I.. Selective Synthesis of Fatty Monoglycerides by Using Functionalized Mesoporous Catalysts Applied Catalysis A: General 254: 173-188 (2003): 173-188.
[29] I. Diaz, C. Marquez-Alvarez, C. MA., Mohino C., M. C.. Combined Alkyl and Sulfonic Acid Functionalization of Mcm-41 Type Silica Part 1. Synthesis and Characterization Journal of Catalysis 193: 283-294 (2000): 283-294.
[30] I.K. Mbaraka, B.H. Shanks. Design of Multifunctionalized Mesoporous Silicas for Esterification of Fatty Acids Journal of Catalysis 229: 365-373 (2005): 365-373.
[31] I.K. Mbaraka, B.H. Shanks. Conversion of Oils and Fats using Advanced Mesoporous Heterogeneous Catalysts Journal of the American Oil Chemists' Society 83: 79 (2006): 79.
[32] I.K. Mbaraka, B.H. Shanks. Acid Strength Variation due to Spatial Location of Organosulfonic Acid Groups on Mesoporous Silicas Journal of Catalysis 244: 78-85 (2006): 78-85.
[33] Y. Tang, S. Miao, S. M., Shanks S.. Applied Catalysis A: General 375: 310-317 (2010): 310-317.
[34] M. Okumura, A. Takagaki, A. T., Toda A., T. A., M. Toda, M. T., Kondo M.. Acid-Catalyzed Reactions on Flexible Polycyclic Aromatic Carbon in Amorphous Carbon Chemistry of Materials 18: 3039-3045 (2006): 3039-3045.
[35] M. Toda, A. Takagaki, A. T., Okamura A., O. A., M. Okamura, M. O.. Green Chemistry: Biodiesel made with Sugar Catalysts Nature 438: 178 (2005): 178.
[36] R. Xing, Y.M. Liu, Y. L., Wang Y.M., W. Y., Y. Wang, Y. W., Chen Y.. Active Solid Acid Catalysts Prepared by Sulfonation of Carbon- ization-Controlled Mesoporous Carbon Materials Microporous and Mesoporous Materials 105: 41-48 (2009): 41-48.
[37] Y. Liu, J. Chen, J. C., Yao J., Y. J., J. Yao. Preparation and Properties of Sulfonated Carbon-Silica Composites from Sucrose Dispersed on MCM-48 Chemical Engineering Science 148: 201-206 (2009): 201-206.
[38] I.K. Mbaraka, D.R. Radu, D. R., Lin D.R.. Organosulfonic Acid Functionalized Mesoporous Silicas for the Esterification of Fatty Acid Journal of Catalysis 219: 329-336 (2003): 329-336.
[39] W.R. Livingston () Biomass Ash Characteristics and Behaviour in Combustion, Gasification and Pyrolysis Systems.
[40] P.R. Patwardhan, J.A. Satrio, J. S., Brown J.A.. Influence of Inorganic Salts and Switchgrass Ash on Primary Pyrolysis Products of Cellulose Bioresource Technology 101: 4646-4655 (2010): 4646-4655.
[41] E.L. Kunkes, D.A. Simonetti, D. S., West D.A., W. D., R.M. West. Catalytic Conversion of Biomass to Monofunctional Hydrocarbons and Targeted Liquid-Fuel Classes Science 322: 417-421 (2008): 417-421.
[42] R.M. West, E.L. Kunkes, E. K., Simonetti E.L.. Catalytic Conversion of Biomass-Derived Carbohydrates to Fuels and Chemicals by Formation and Upgrading of Mono-Functional Hydrocarbon Intermediates Catalysis Today 147: 115-125 (2009): 115-125.
[43] Y. Roman-Leshkov, J.N. Chheda, J. C.. Phase Modifiers Promote Efficient Production of Hydroxymethylfurfural from Fructose Science 312: 1933-1937 (2006): 1933-1937.
[44] J.N. Chheda, J.A. Dumesic. An Overview of Dehydration, Aldol-Con- densation and Hydrogenation Processes for Production of Liquid Alkanes from Biomass-Derived Carbohydrates Catalysis Today 123: 59-70 (2007): 59-70.
[45] J.N. Chheda, G.W. Huber, G. H.. Liquid-Phase Catalytic Processing of Biomass to Fuels and Chemicals Angewandte Chemie International Edition 46: 7164-7183 (2007): 7164-7183.
[46] C.A. Gaertner, J.C. Serrano-Ruiz, J. SR., Braden J.C.. Catalytic Coupling of Carboxylic Acids by Ketonization as a Processing Step in Biomass Conversion Journal of Catalysis 266: 71-78 (2009): 71-78.
[47] D.A. Simonetti, J.A. Dumesic. Catalytic Strategies for Changing the Energy Content and Achieving C–C Coupling in Biomass-Derived Oxygenated Hydrocarbons ChemSusChem 1: 725-733 (2008): 725-733.
[48] R.M. West, Z.Y. Liu, Z. L., Peter Z.Y., P. Z.. Carbon-Carbon Bond Formation for Biomass-Derived Furfurals and Ketones by Aldol Condensation in a Biphasic System Journal of Molecular Catalysis A: Chemical 296: 18-27 (2008): 18-27.
[49] J.N. Chheda, R. Roman-Leshkov, R. RL.. Production of 5-Hy- droxymethylfurfural and Furfural by Dehydration of Biomass-Derived Mono- and Poly-Saccharides Green Chemistry 9: 342-350 (2007): 342-350.
[50] R. Roman-Leshkov, J.A. Dumesic. Solvent Effects on Fructose Dehydration to 5-Hydroxymethylfurfural in Biphasic Systems Saturated with Inorganic Salts Topics in Catalysis 52: 297-303 (2009): 297-303.